
Differential Interferon Signaling Regulation and Oxidative Stress Responses in the Cerebral Cortex and Cerebellum Could Account for the Spatiotemporal Pattern of Neurodegeneration in Niemann–Pick Disease Type C
 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Transcriptome Analysis
2.3. Data Pre-Processing
2.4. Differential Gene Expression Analysis
2.5. Pathway Enrichment
2.6. Gene Set Enrichment Analysis
3. Results
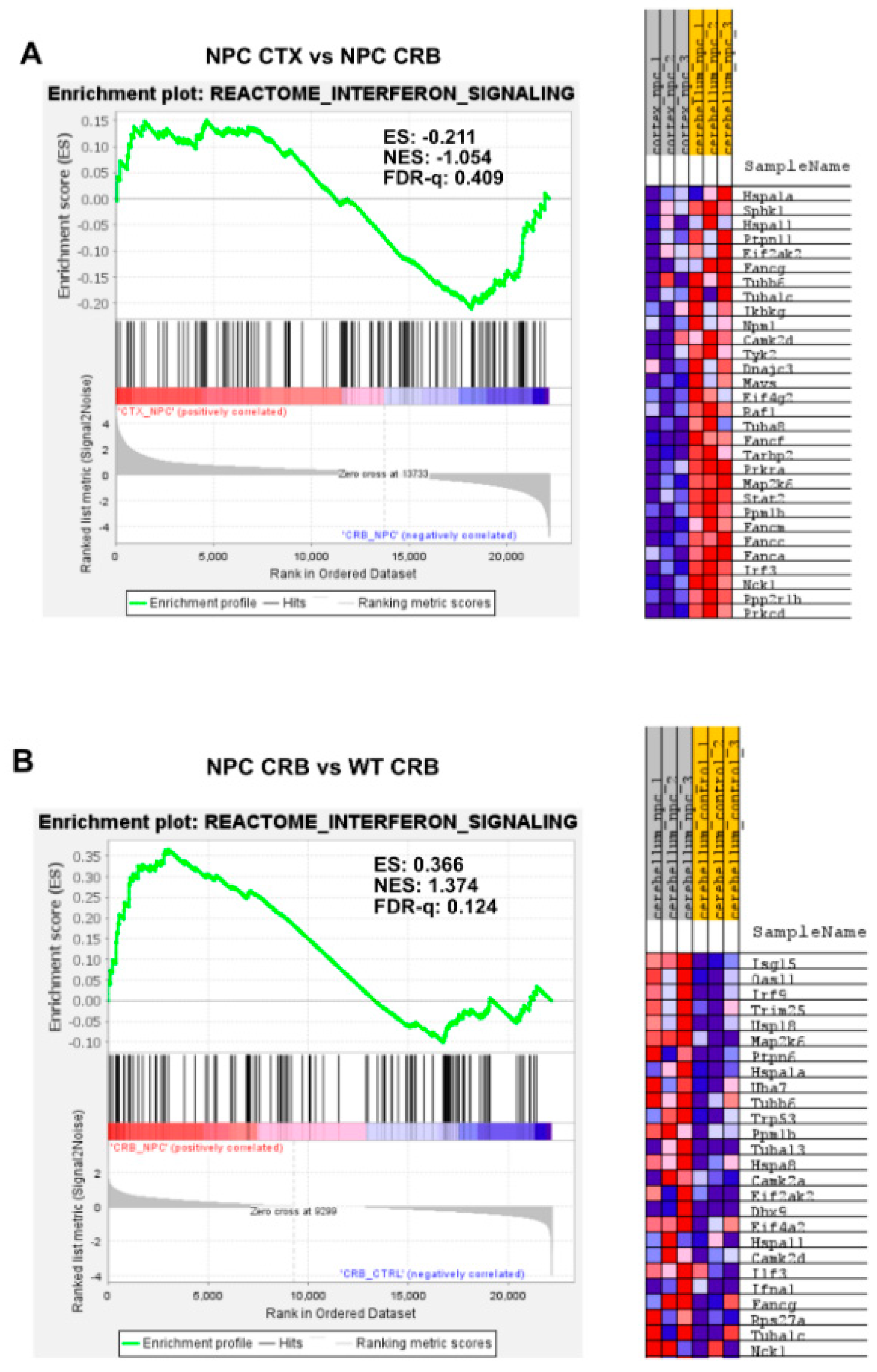
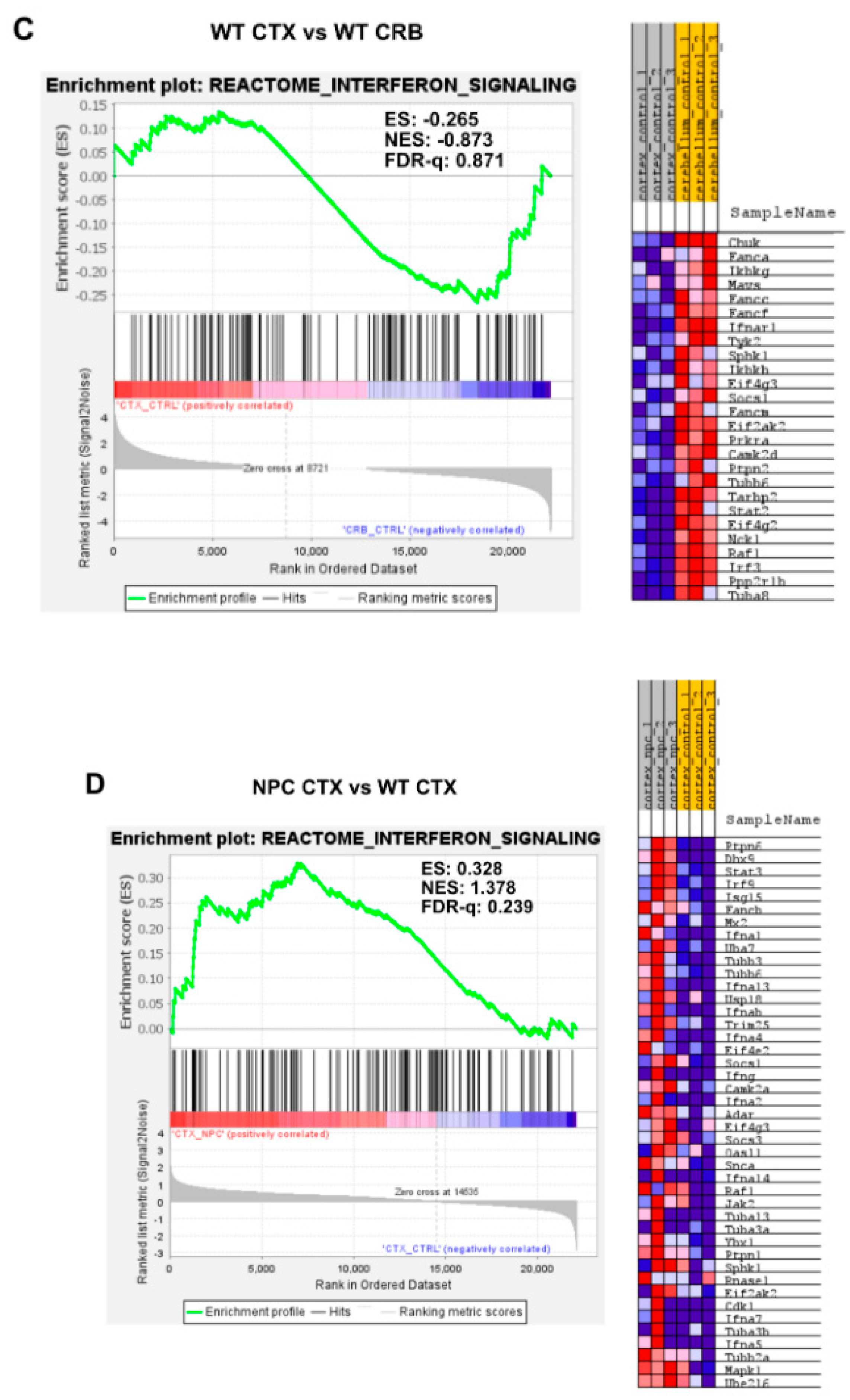
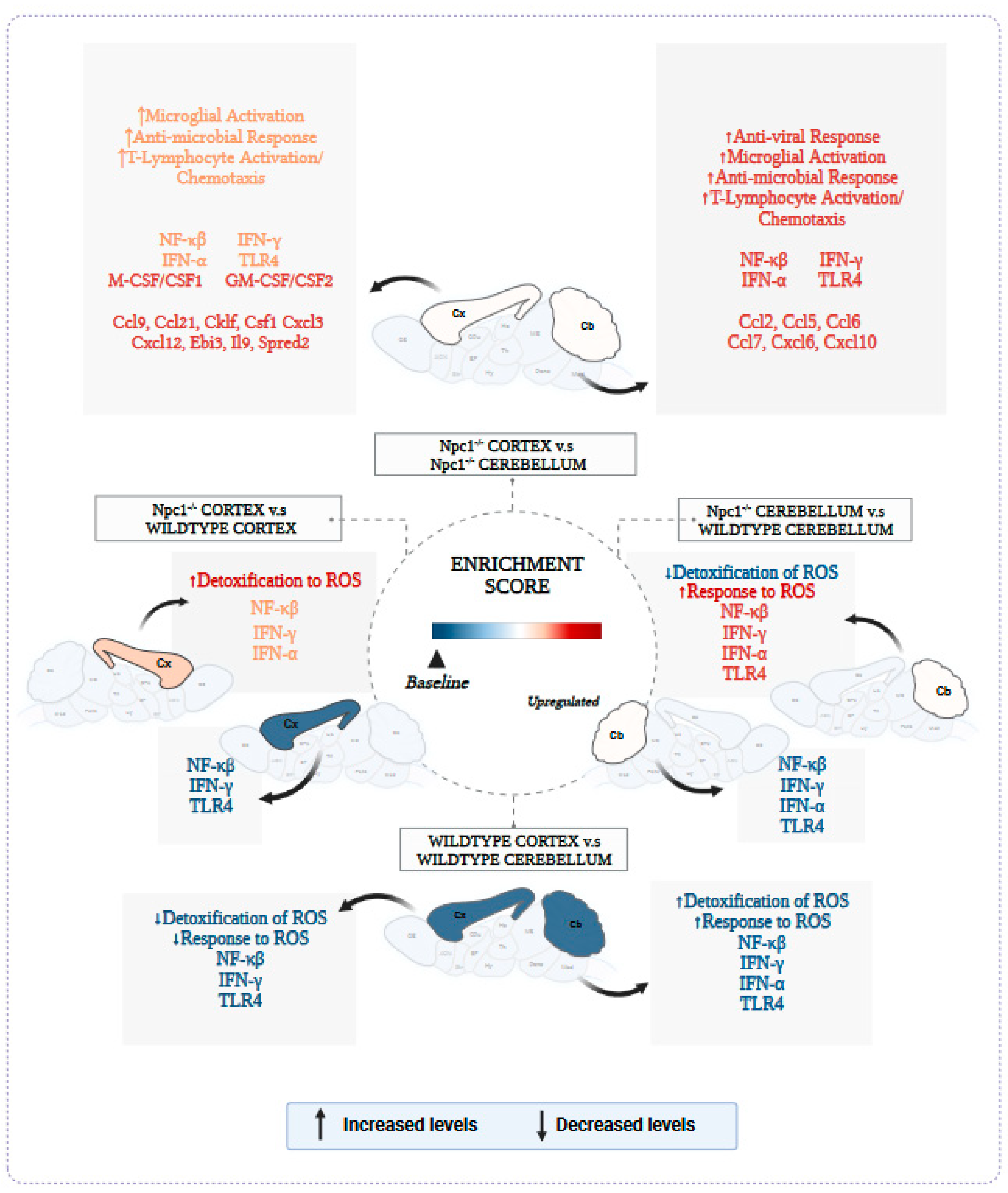
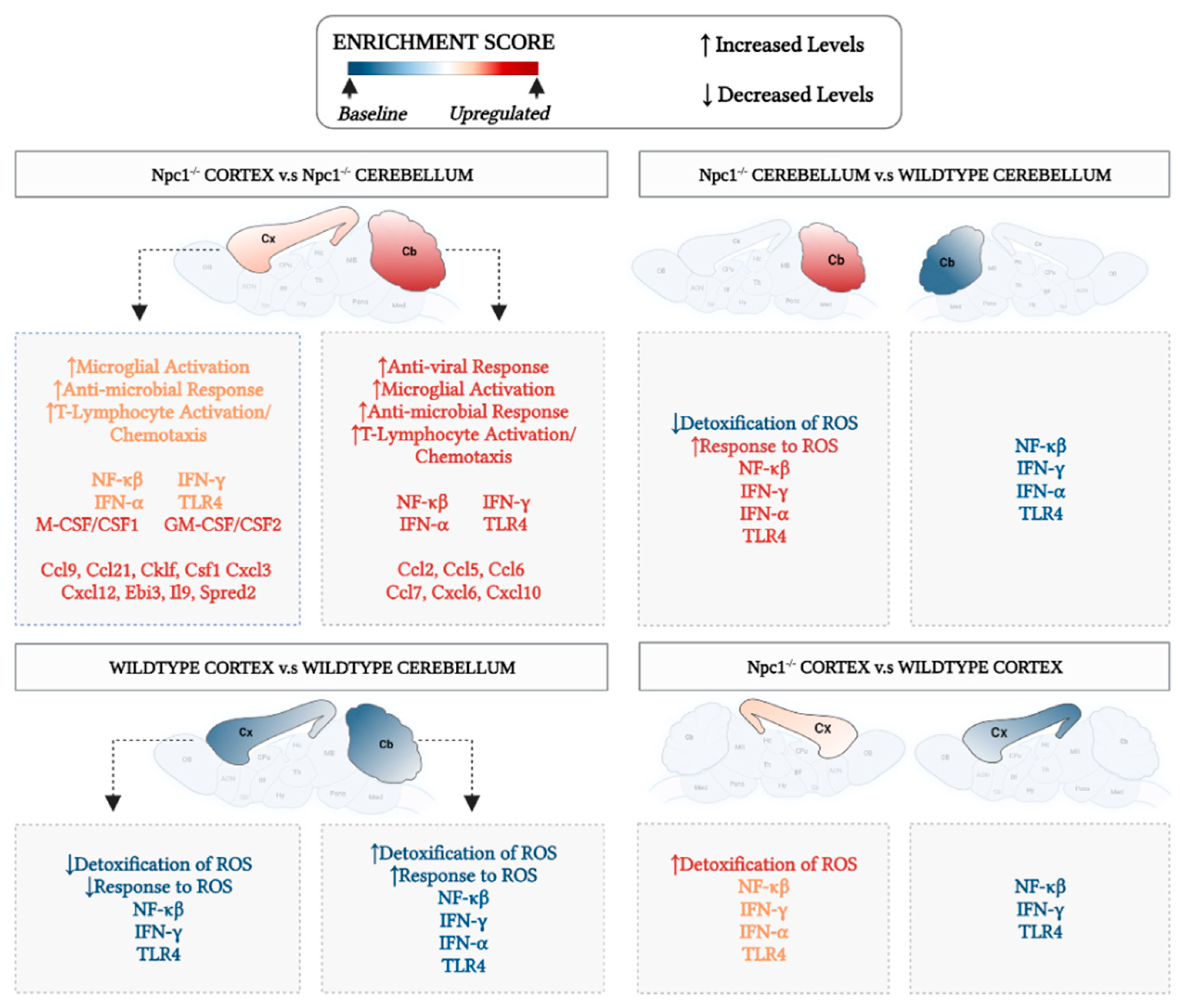
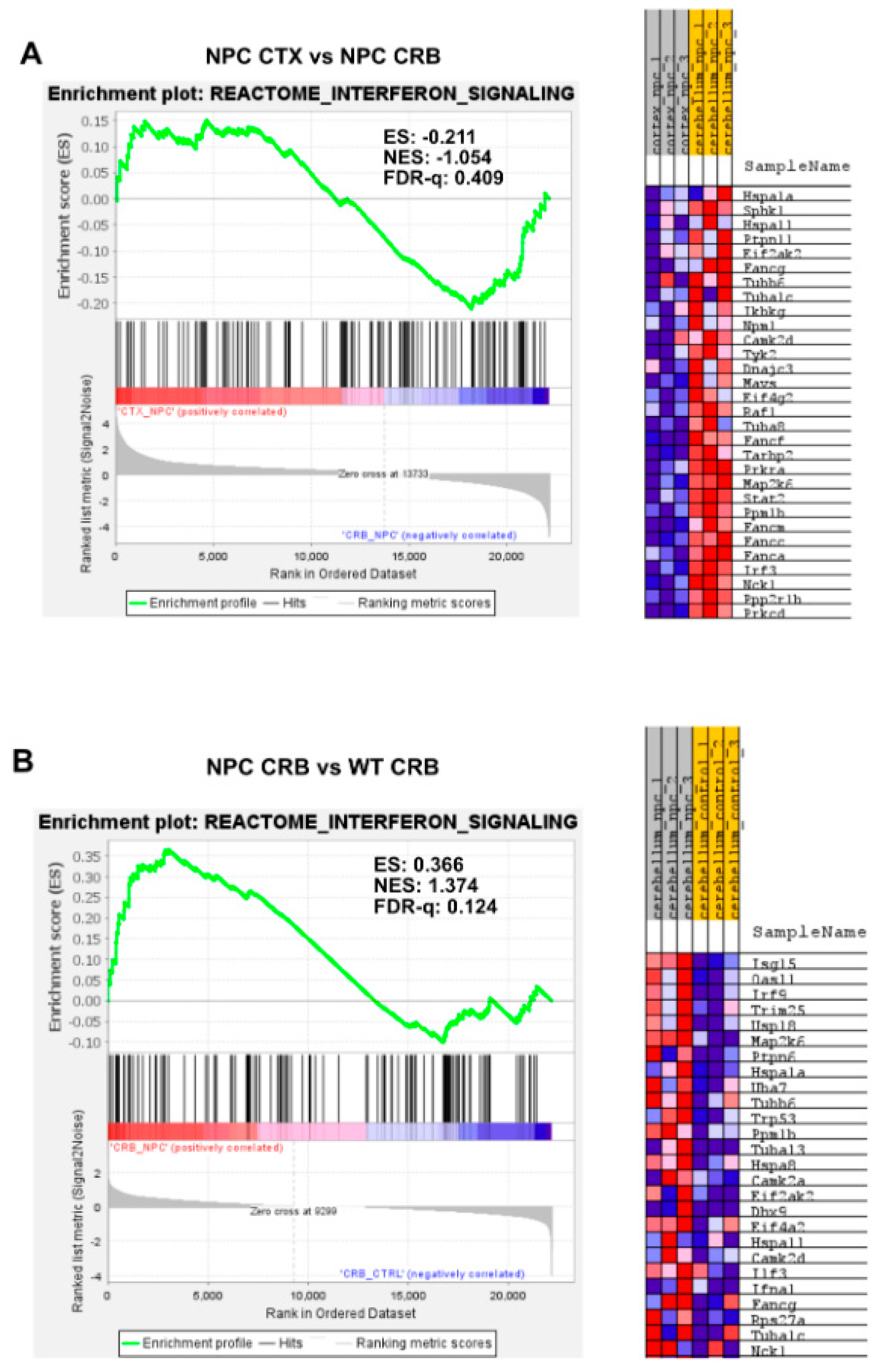
3.1. Interferon Signaling Is Higher in the Cerebellum Than in the Cerebral Cortex in NPC
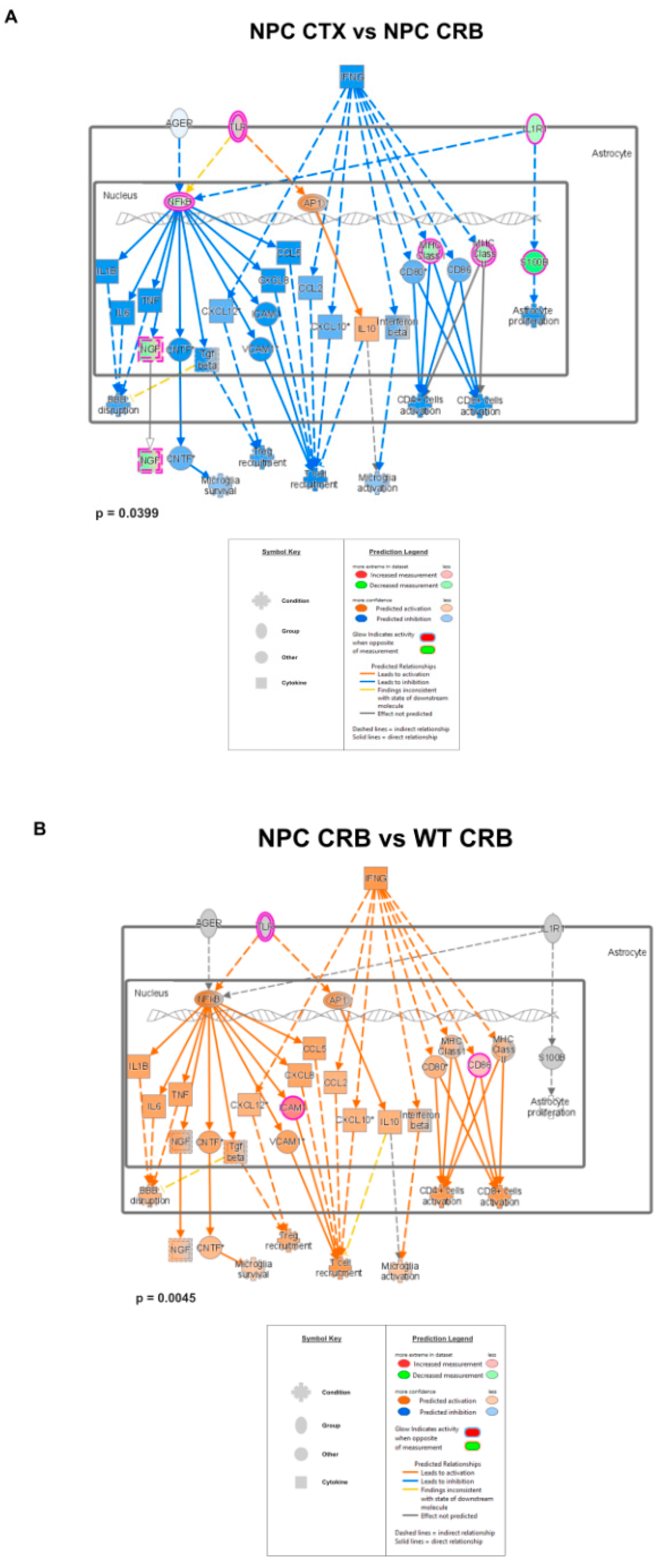
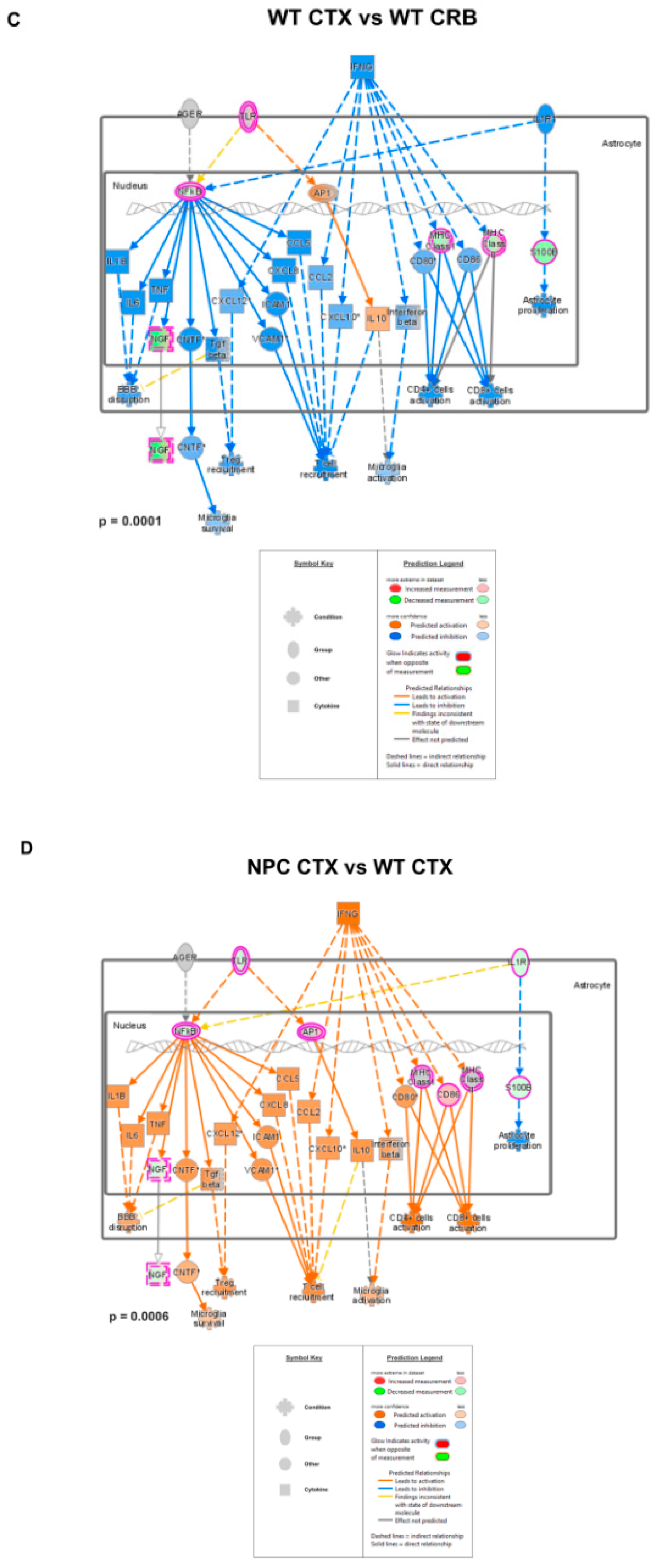
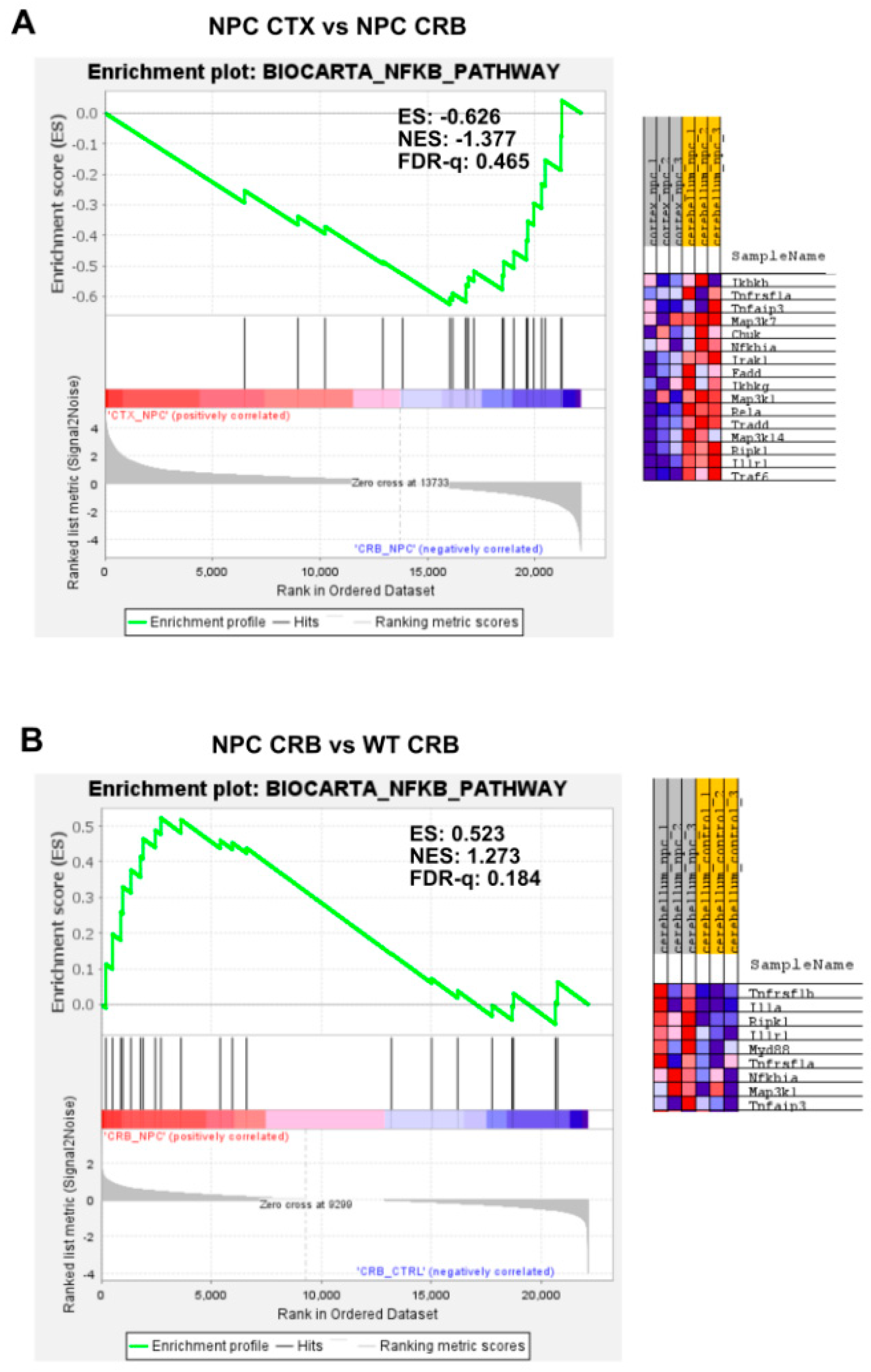
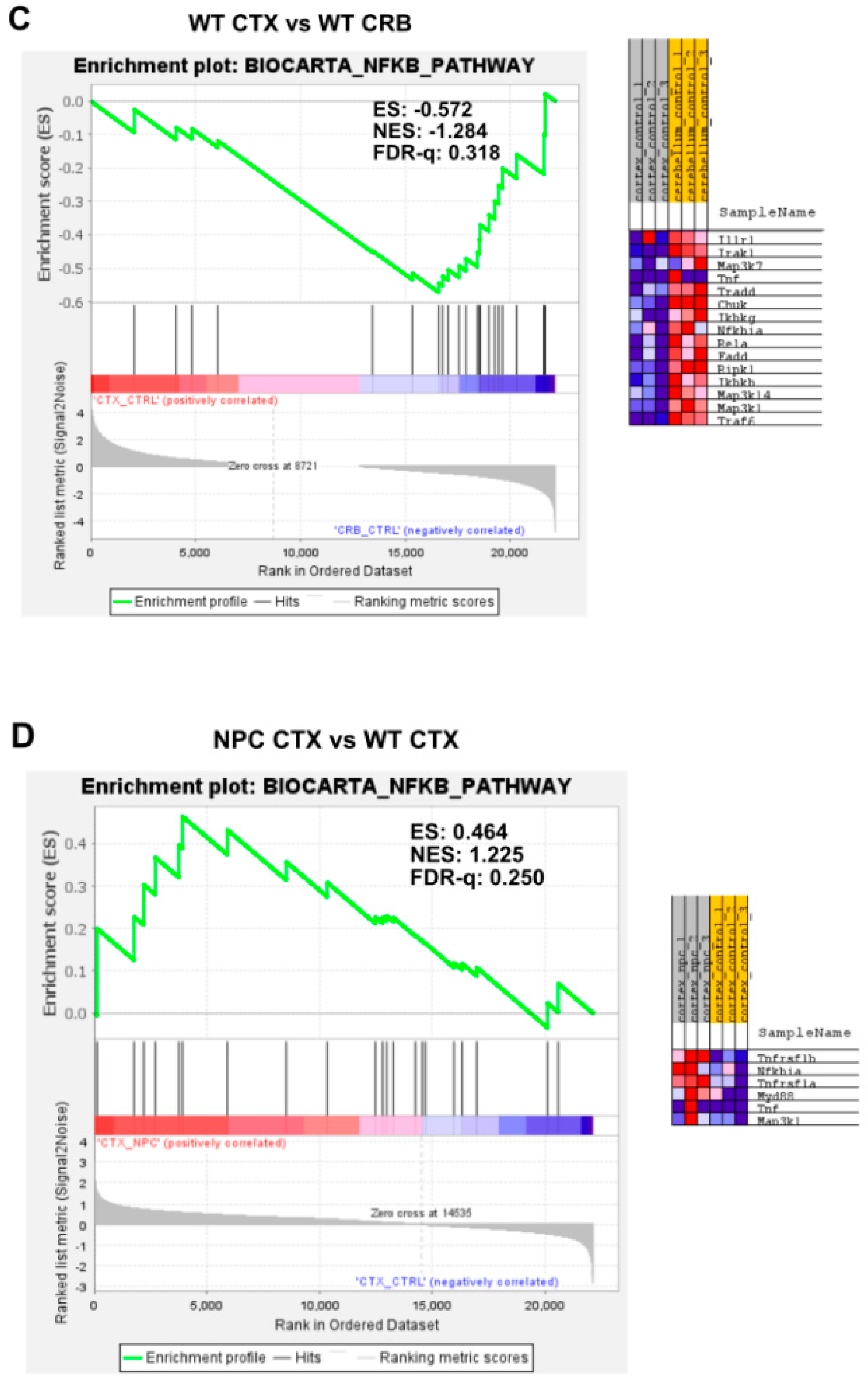
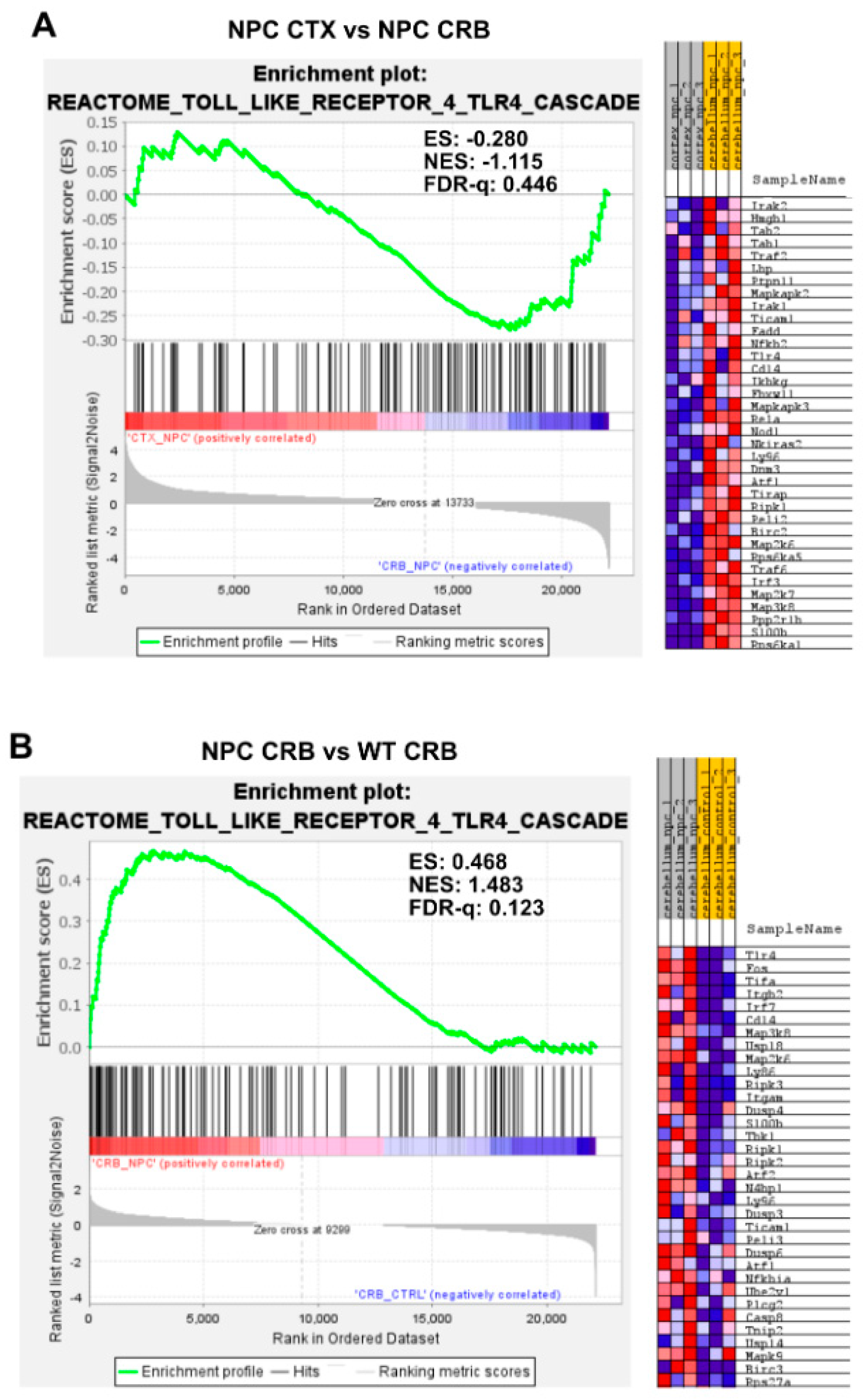
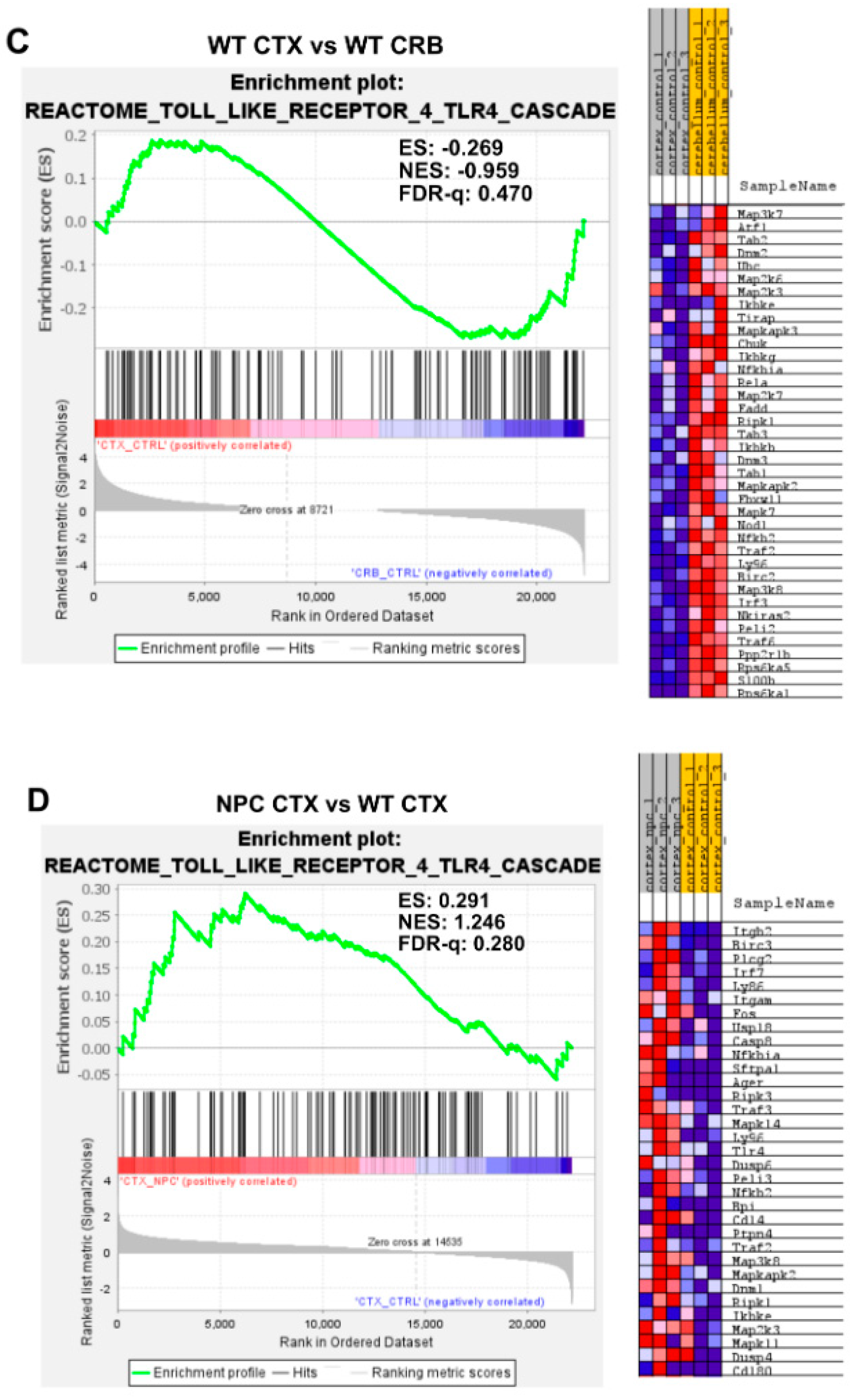
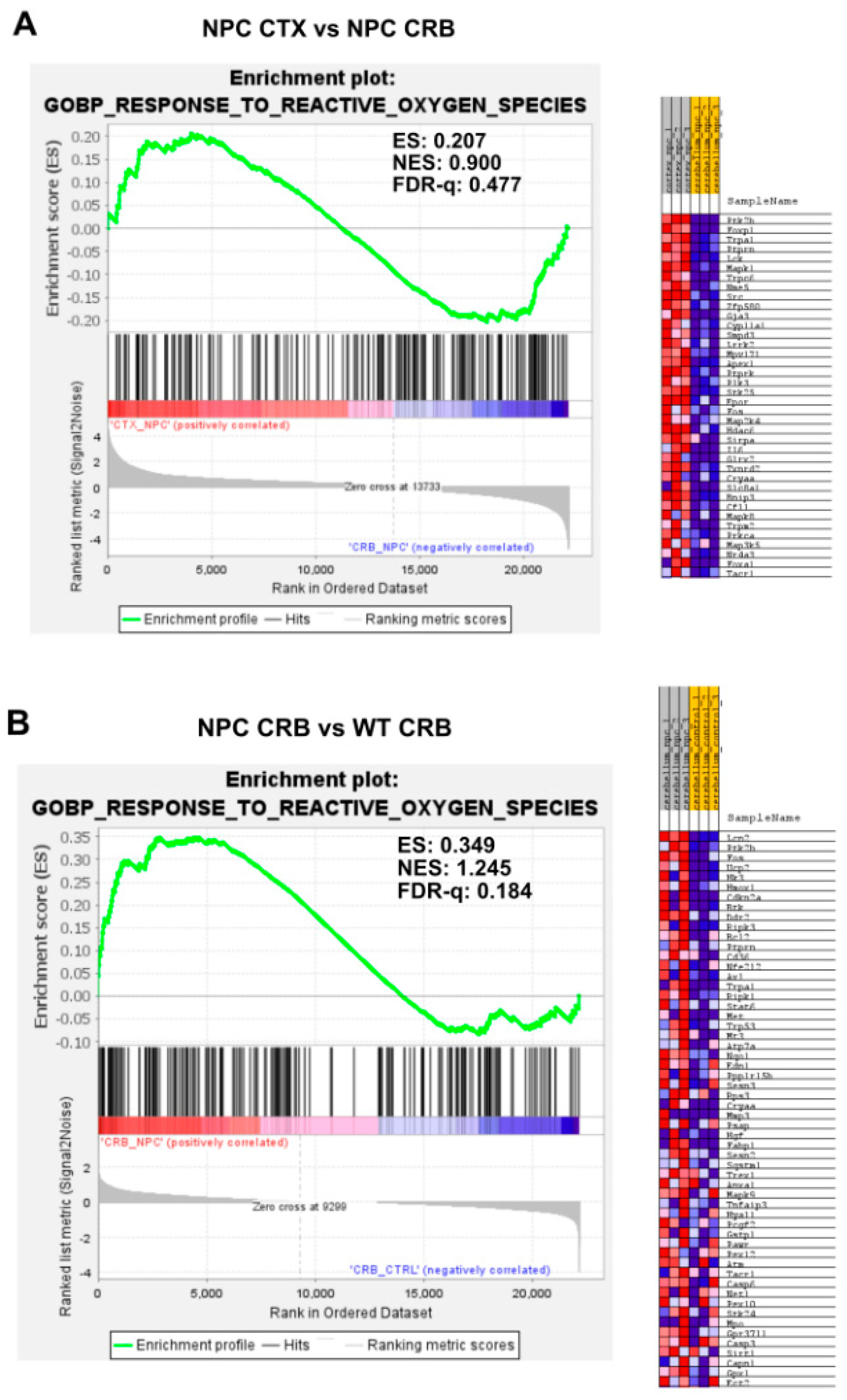
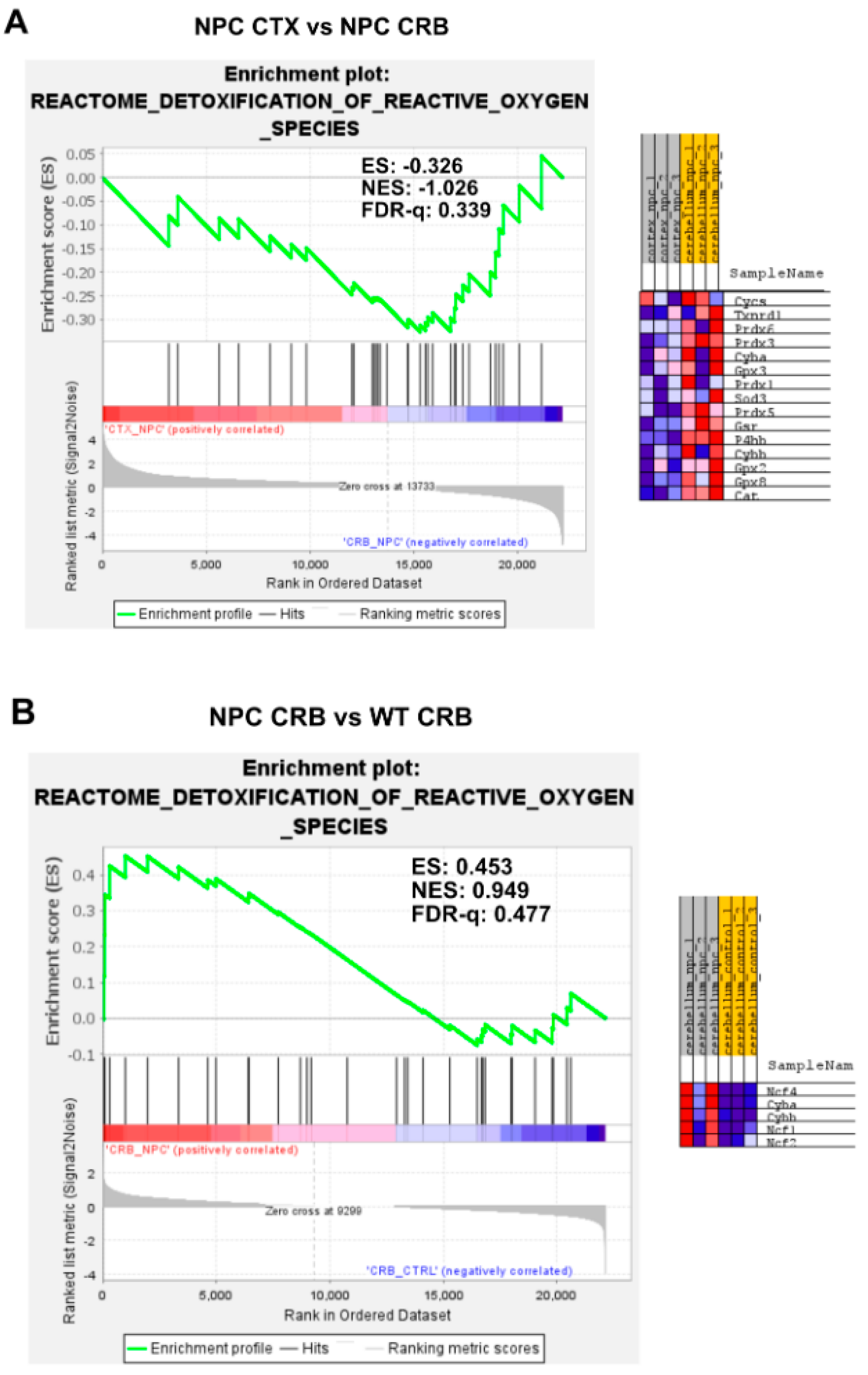
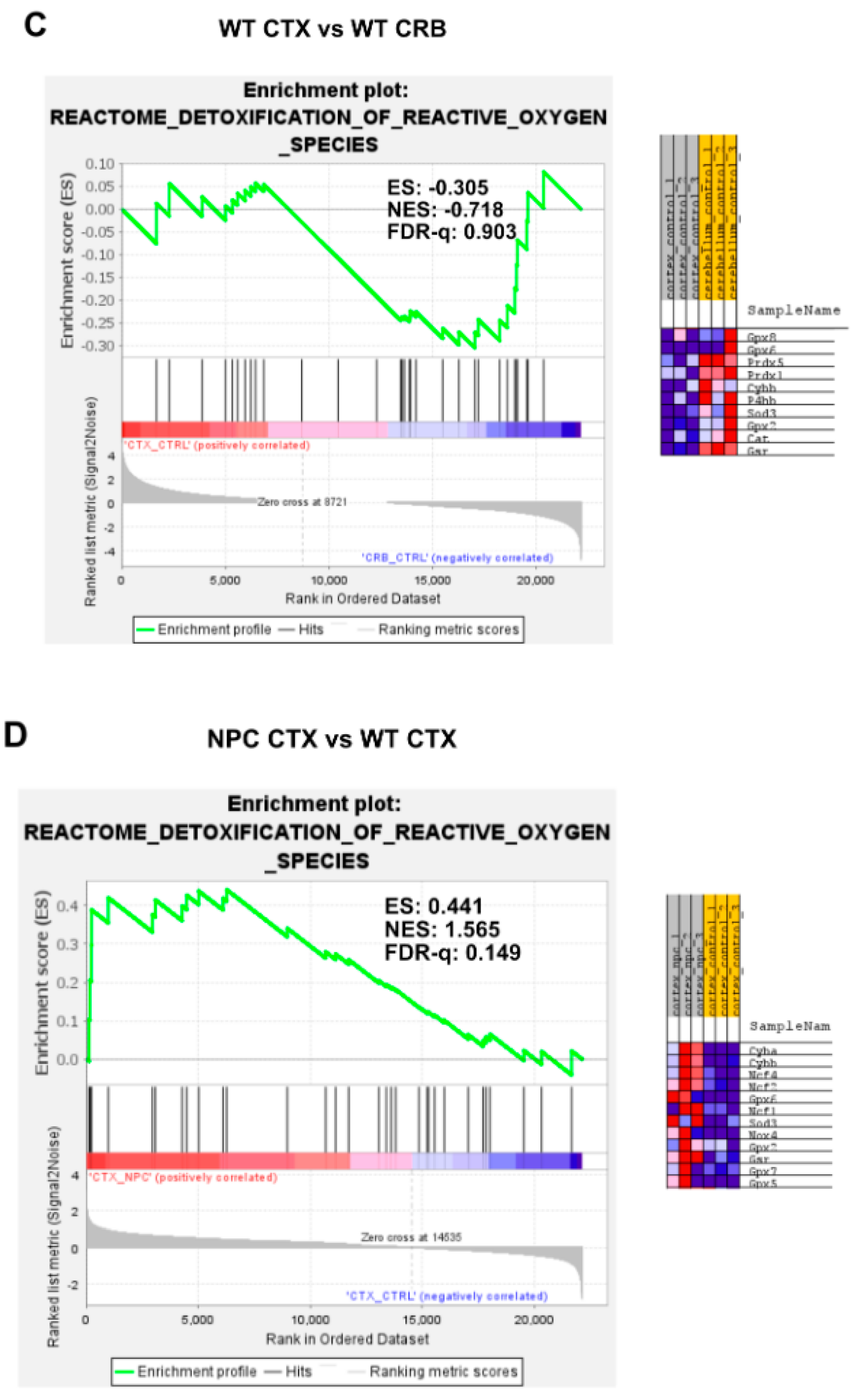
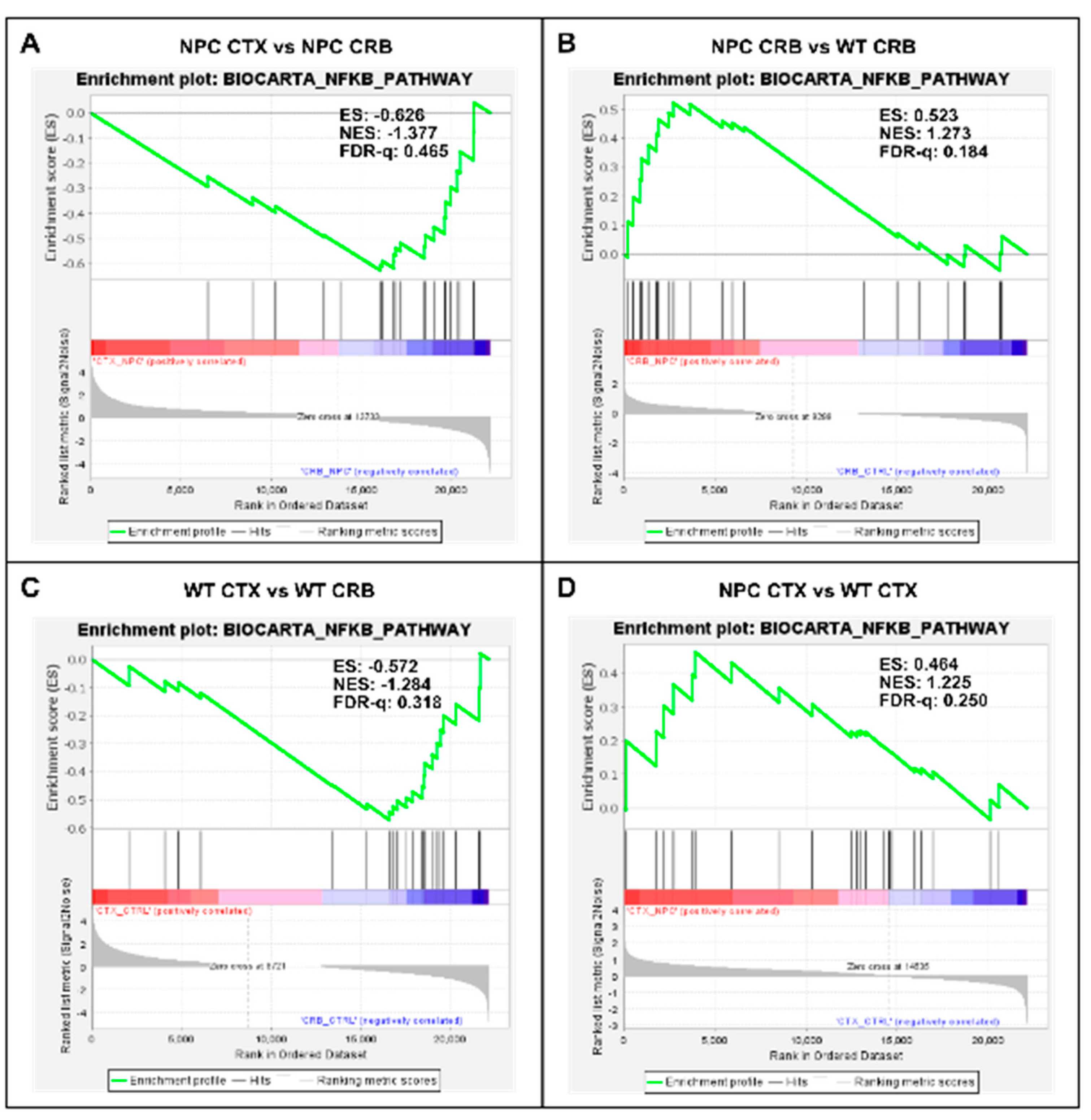
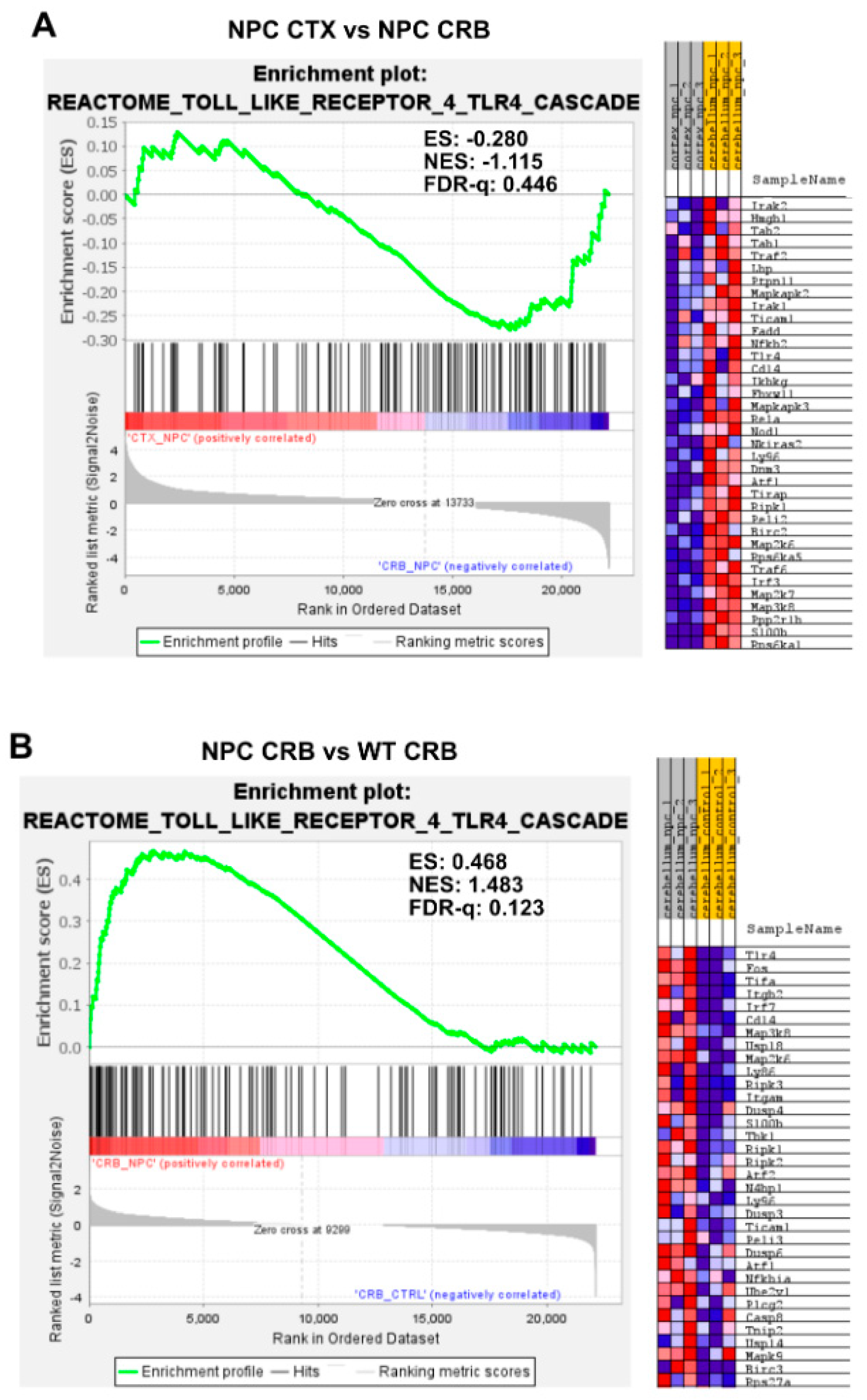
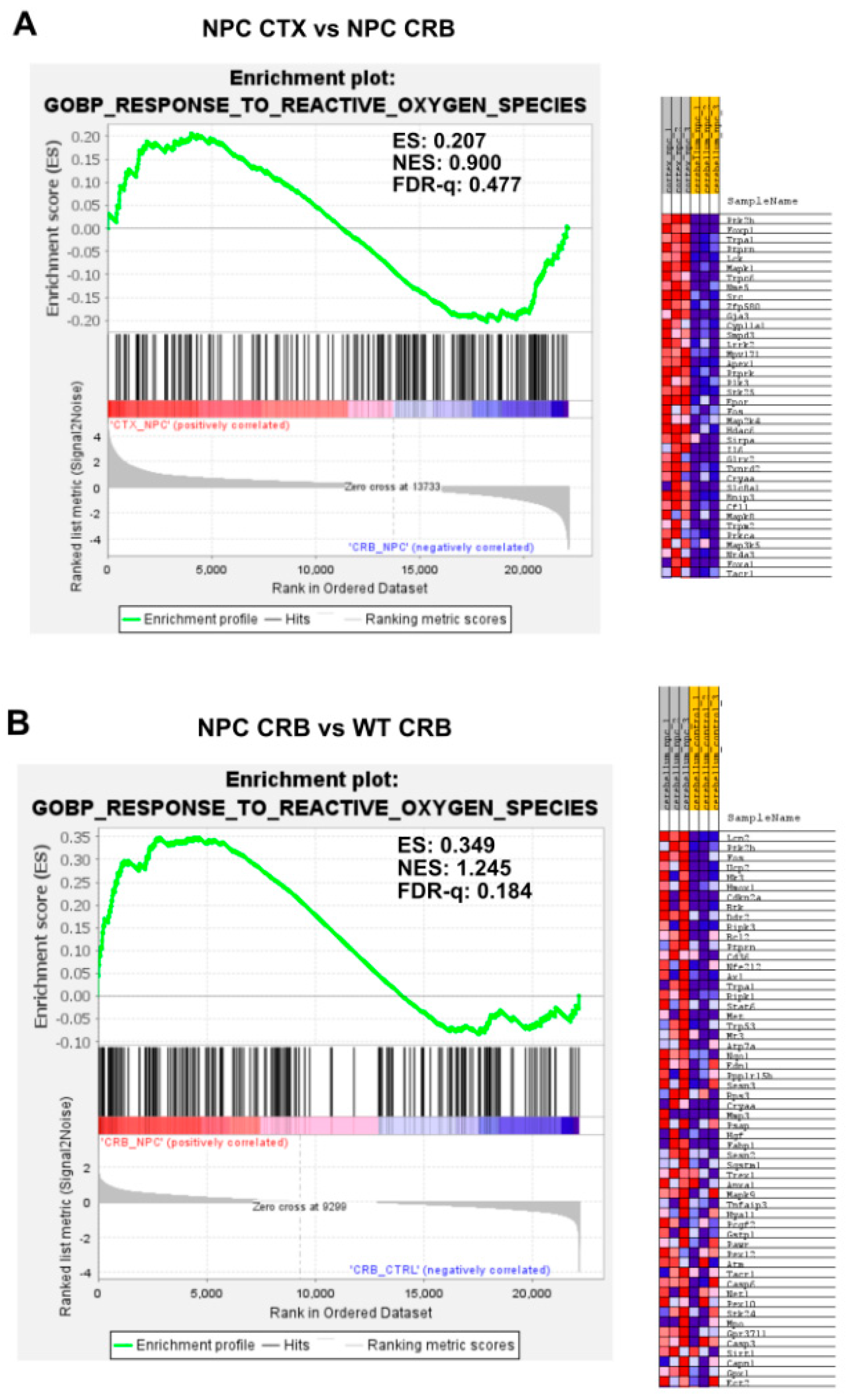
3.2. Inflammatory Pathways Linked to DAMP Generation, Excessive ROS, and Lipid Peroxidation in the Npc1−/− Brain Are More Active in the Cerebellum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A. Expanded Figures to Enhance Readability of Figure 1, Figure 2, Figure 3, Figure 4, Figure 5 and Figure 6. Figure 1 and Figure 3, Figure 4, Figure 5 and Figure 6 Include Heatmaps of Core Enriched Genes for Each Comparison Panel

References
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef]
- Nunes, A.; Pressey, S.N.; Cooper, J.D.; Soriano, S. Loss of amyloid precursor protein in a mouse model of Niemann-Pick type C disease exacerbates its phenotype and disrupts tau homeostasis. Neurobiol. Dis. 2011, 42, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Kodachi, T.; Matsumoto, S.; Mizuguchi, M.; Osaka, H.; Kanai, N.; Nanba, E.; Ohno, K.; Yamagata, T. Severe demyelination in a patient with a late infantile form of Niemann-Pick disease type C. Neuropathology 2017, 37, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Evans, E.; Platt, F.M. Lipids on trial: The search for the offending metabolite in Niemann-Pick type C disease. Traffic 2010, 11, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal cal cium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef]
- Goicoechea, L.; de la Rosa, L.C.; Torres, S.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial cholesterol: Metabolism and impact on redox biology and disease. Redox. Biol. 2023, 61, 102643. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.D.; Shin, A.; Mayagoitia, K.; Siebold, L.; Rubini, M.; Wilson, C.G.; Bellinger, D.L.; Soriano, S. Loss of amyloid precursor protein exacerbates early inflammation in Niemann-Pick disease type C. J. Neuroinflamm. 2019, 16, 269. [Google Scholar] [CrossRef]
- Shin, S.D.K.; Shin, A.; Mayagoitia, K.; Wilson, C.G.; Bellinger, D.L.; Soriano, S. Interferon downstream signaling is activated early in pre-symptomatic Niemann-Pick disease type C. Neurosci. Lett. 2019, 706, 43–50. [Google Scholar] [CrossRef]
- Baudry, M.; Yao, Y.; Simmons, D.; Liu, J.; Bi, X. Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: Immunohistochem ical observations of microglia and astroglia. Exp. Neurol. 2003, 184, 887–903. [Google Scholar] [CrossRef]
- Pressey, S.N.; Smith, D.A.; Wong, A.M.; Platt, F.M.; Cooper, J.D. Early glial activation, synaptic changes and axonal pathology in the thalamocortical system of Niemann-Pick type C1 mice. Neurobiol. Dis. 2012, 45, 1086–1100. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Chasset, F.; Dayer, J.M.; Chizzolini, C. Type I Interferons in Systemic Autoimmune Diseases: Distinguishing between Af ferent and Efferent Functions for Precision Medicine and Individualized Treatment. Front. Pharmacol. 2021, 12, 633821. [Google Scholar] [CrossRef] [PubMed]
- Choubey, D.; Moudgil, K.D. Interferons in autoimmune and inflammatory diseases: Regulation and roles. J. Interferon Cytokine Res. 2011, 31, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondial dehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, S.; Mellon, S.H.; Butters, T.D.; Nevyjel, M.; Covey, D.F.; Bembi, B.; Dardis, A. Oxidative stress in NPC1 deficient cells: Protective effect of allopregnanolone. J. Cell Mol. Med. 2009, 13, 3786–3796. [Google Scholar] [CrossRef] [PubMed]
- Kielian, T. Lysosomal storage disorders: Pathology within the lysosome and beyond. J. Neurochem. 2019, 148, 568–572. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Suzuki, M.; Sugimoto, Y.; Ohsaki, Y.; Ueno, M.; Kato, S.; Kitamura, Y.; Hosokawa, H.; Davies, J.P.; Ioannou, Y.A.; Vanier, M.T.; et al. Endosomal accumulation of Toll-like receptor 4 causes constitutive secretion of cytokines and activation of signal transducers and activators of transcription in Niemann-Pick disease type C (NPC) fibroblasts: A potential basis for glial cell activa tion in the NPC brain. J. Neurosci. 2007, 27, 1879–1891. [Google Scholar]
- Patterson, M.C.; Hendriksz, C.J.; Walterfang, M.; Sedel, F.; Vanier, M.T.; Wijburg, F. Recommendations for the diagnosis and management of Niemann-Pick disease type C: An update. Mol. Genet. Metab. 2012, 106, 330–344. [Google Scholar] [CrossRef]
- Patterson, M.C.; Mengel, E.; Wijburg, F.A.; Muller, A.; Schwierin, B.; Drevon, H.; Vanier, M.T.; Pineda, M. Disease and patient characteristics in NP-C patients: Findings from an international disease registry. Or. Phanet J. Rare Dis. 2013, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Murayama, S.; Pentchev, P.G.; Suzuki, K. Cerebellar degeneration in the Niemann-Pick type C mouse. Acta Neuropathol. 1993, 85, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Ko, D.C.; Milenkovic, L.; Beier, S.M.; Manuel, H.; Buchanan, J.; Scott, M.P. Cell-autonomous death of cerebellar purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet. 2005, 1, 81–95. [Google Scholar] [PubMed]
- German, D.C.; Quintero, E.M.; Liang, C.L.; Ng, B.; Punia, S.; Xie, C.; Dietschy, J.M. Selective neurodegeneration, without neurofibrillary tangles, in a mouse model of Niemann-Pick C disease. J. Comp. Neurol. 2001, 433, 415–425. [Google Scholar] [CrossRef]
- Herbert, A.S.; Davidson, C.; Kuehne, A.I.; Bakken, R.; Braigen, S.Z.; Gunn, K.E.; Whelan, S.P.; Brummelkamp, T.R.; Twenhafel, N.A.; Chandran, K.; et al. Niemann-pick C1 is essential for ebolavirus replication and pathogenesis in viv. Mbio 2015, 6, e00565-15. [Google Scholar] [CrossRef]
- Côté, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 2011, 477, 344–348. [Google Scholar] [CrossRef]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef]
- Liu, M.; Guo, S.; Hibbert, J.M.; Jain, V.; Singh, N.; Wilson, N.O.; Stiles, J.K. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Fac. Tor Rev. 2011, 22, 121–130. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upstream Regulator | Npc1−/− vs. Npc1+/+ | ||
|---|---|---|---|
| Z-Score | p-Overlap | # Target | |
| IFN-γ | 4.104 | 5.12 × 10−11 | 84 |
| M-CSF/CSF1 | 3.785 | 6.17 × 10−6 | 21 |
| GM-CSF/CSF2 | 3.598 | 1.00 × 10−5 | 33 |
| IL-33 | 3.036 | 1.22 × 10−3 | 17 |
| TNFα | 2.908 | 9.37 × 10−9 | 96 |
| IL-4 | 2.902 | 1.68 × 10−6 | 57 |
| IL-6 | 2.737 | 4.26 × 10−6 | 46 |
| IL-3 | 2.460 | 3.66 × 10−3 | 21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolan, A.J.; Sanchez, K.L.; Shin, S.D.; White, J.B.; Currais, A.; Soriano-Castell, D.; Wilson, C.G.; Maher, P.; Soriano, S. Differential Interferon Signaling Regulation and Oxidative Stress Responses in the Cerebral Cortex and Cerebellum Could Account for the Spatiotemporal Pattern of Neurodegeneration in Niemann–Pick Disease Type C. Genes 2024, 15, 101. https://doi.org/10.3390/genes15010101
Tolan AJ, Sanchez KL, Shin SD, White JB, Currais A, Soriano-Castell D, Wilson CG, Maher P, Soriano S. Differential Interferon Signaling Regulation and Oxidative Stress Responses in the Cerebral Cortex and Cerebellum Could Account for the Spatiotemporal Pattern of Neurodegeneration in Niemann–Pick Disease Type C. Genes. 2024; 15(1):101. https://doi.org/10.3390/genes15010101
Chicago/Turabian StyleTolan, Andrew J., Kayla L. Sanchez, Samuel D. Shin, Jacob B. White, Antonio Currais, David Soriano-Castell, Christopher G. Wilson, Pamela Maher, and Salvador Soriano. 2024. "Differential Interferon Signaling Regulation and Oxidative Stress Responses in the Cerebral Cortex and Cerebellum Could Account for the Spatiotemporal Pattern of Neurodegeneration in Niemann–Pick Disease Type C" Genes 15, no. 1: 101. https://doi.org/10.3390/genes15010101
APA StyleTolan, A. J., Sanchez, K. L., Shin, S. D., White, J. B., Currais, A., Soriano-Castell, D., Wilson, C. G., Maher, P., & Soriano, S. (2024). Differential Interferon Signaling Regulation and Oxidative Stress Responses in the Cerebral Cortex and Cerebellum Could Account for the Spatiotemporal Pattern of Neurodegeneration in Niemann–Pick Disease Type C. Genes, 15(1), 101. https://doi.org/10.3390/genes15010101