
A Splicing Variant in RDH8 Is Associated with Autosomal Recessive Stargardt Macular Dystrophy

 , ,
, , 
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Whole-Exome Sequencing and Bioinformatics Analysis
2.3. Variant Validation via Sanger Sequencing
2.4. Effect Evaluation of Variant
3. Results
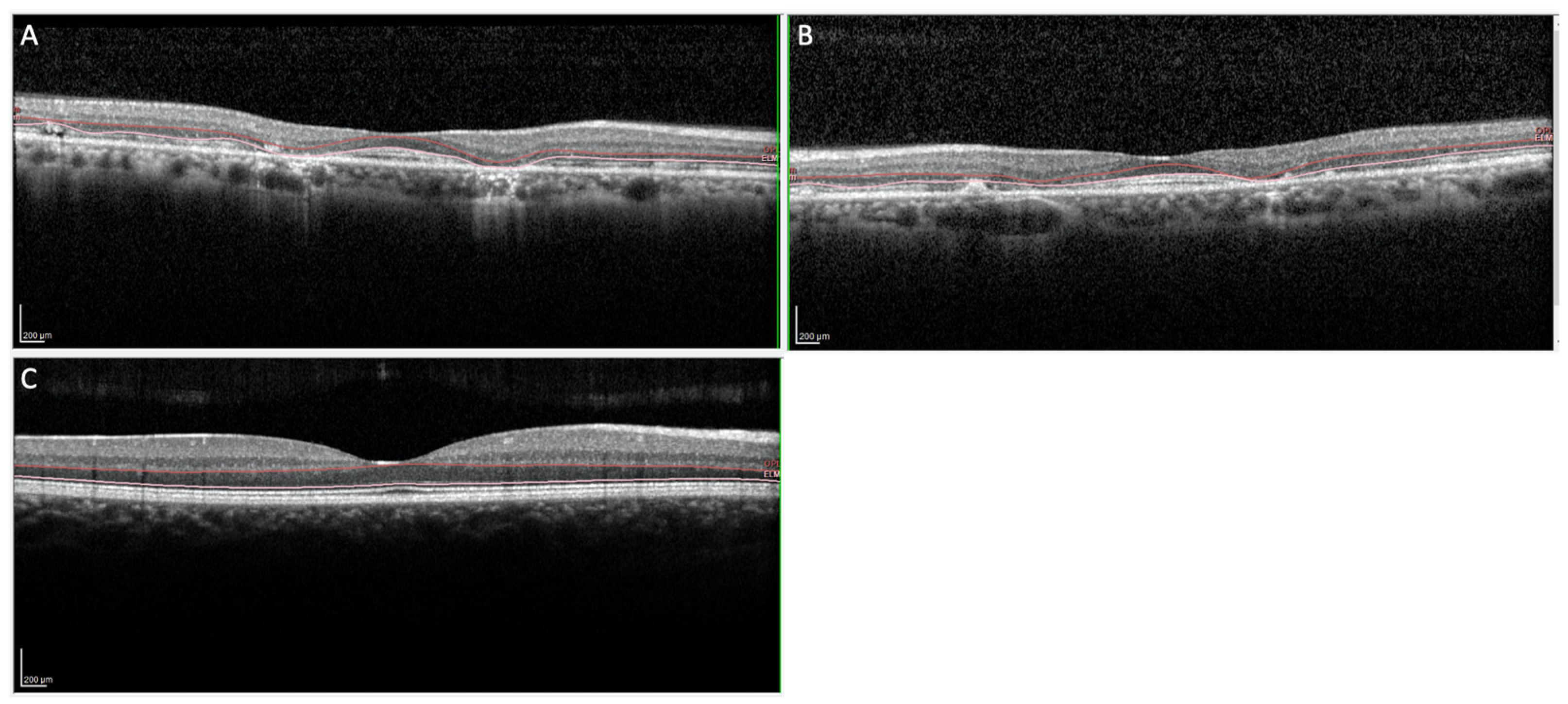
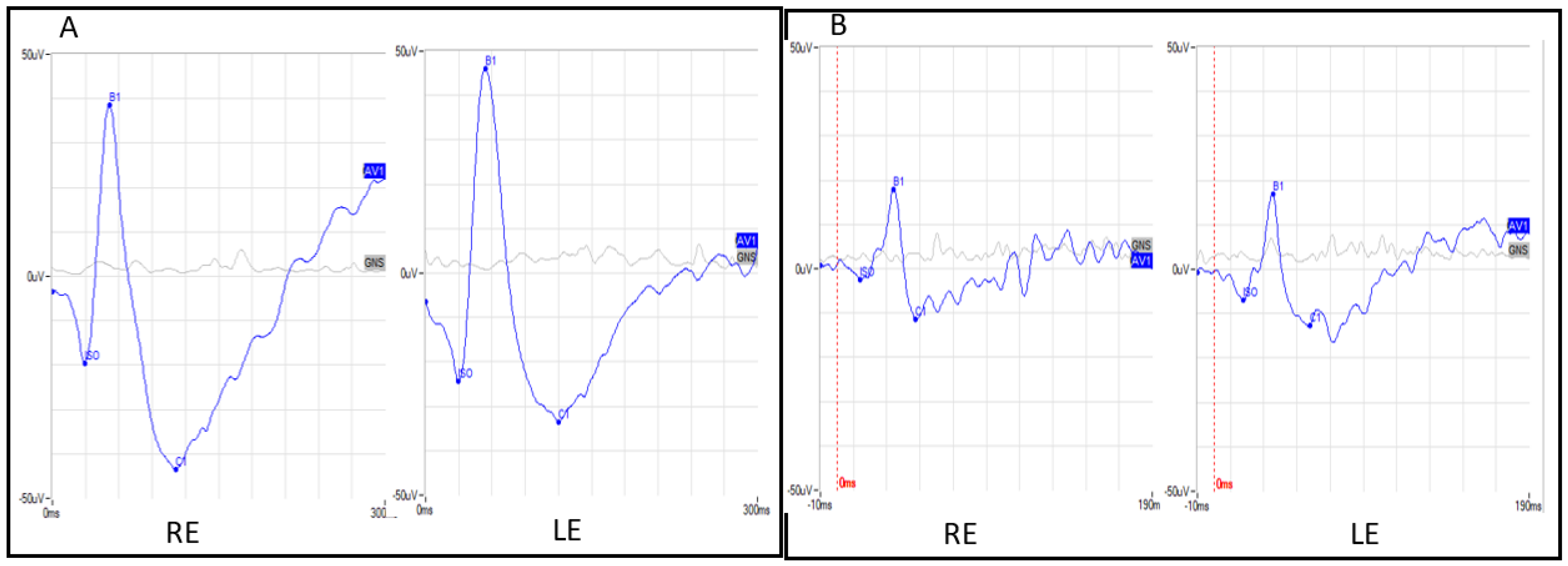
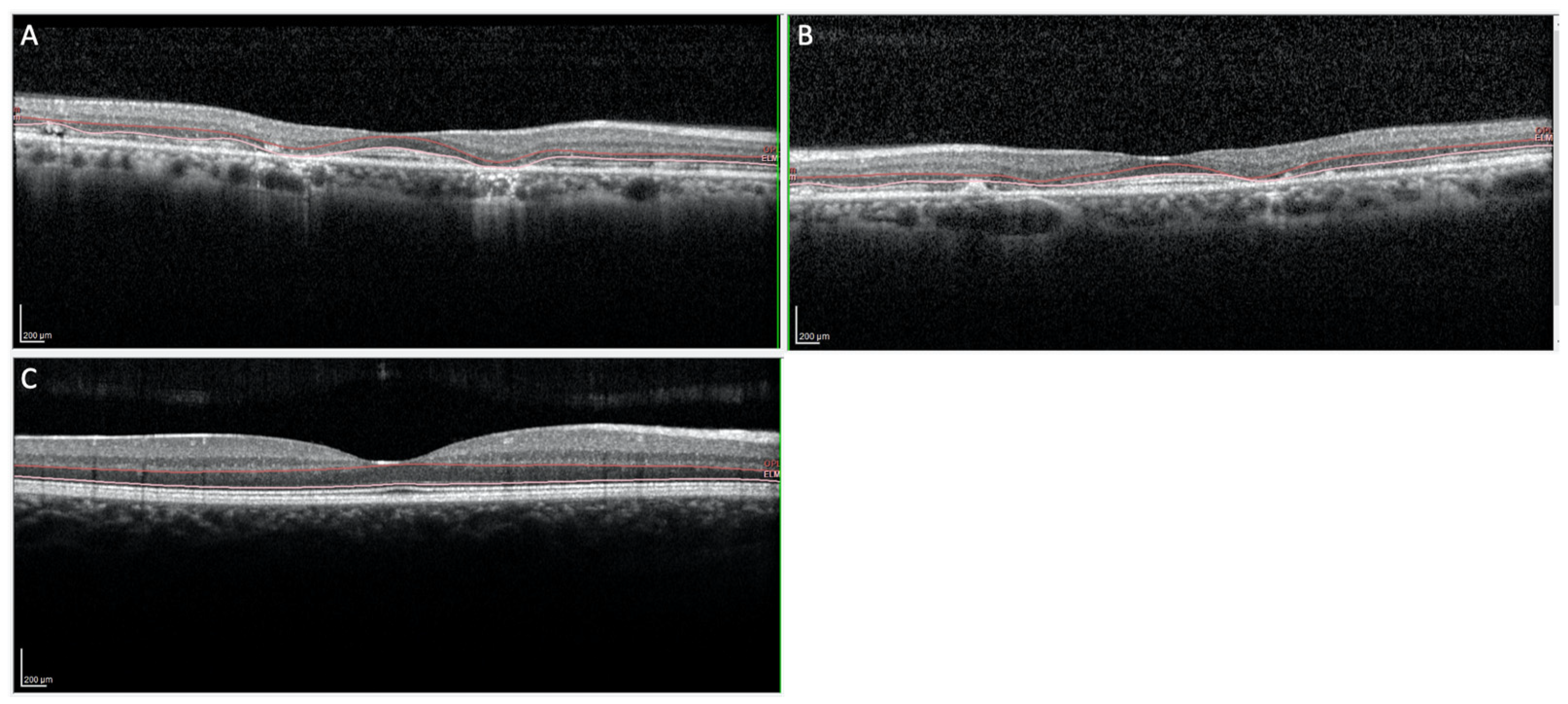
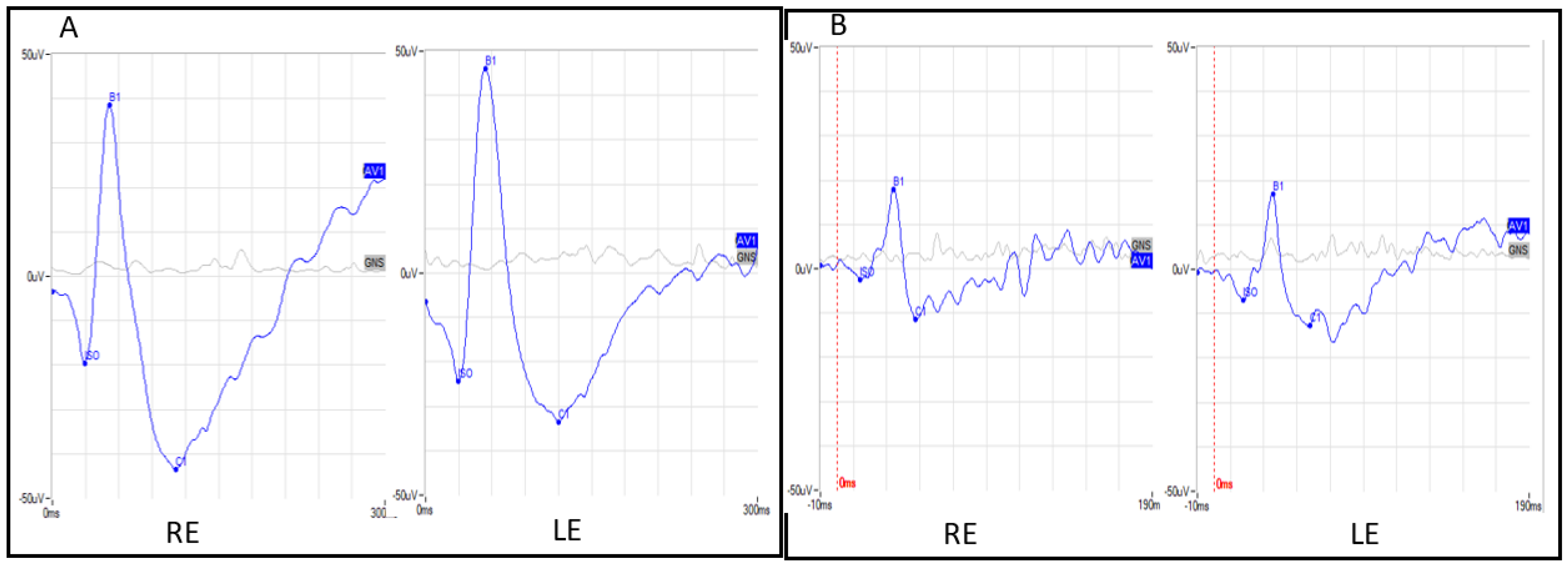
3.1. Clinical Features
3.2. Bioinformatics and Molecular Analyses
3.3. Effect Evaluation of Variant
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stargardt, K. Über familiäre, progressive Degeneration in der Maculagegend des Auges. Albrecht Von Graefes Arch. Klin Ophthalmol. 1909, 71, 534–550. [Google Scholar] [CrossRef]
- Franceschetti, A. A special form of tapetoretinal degeneration: Fundus flavimaculatus. Trans. Am. Acad. Ophthalmol. Otolaryngol. 1965, 69, 1048–1053. [Google Scholar] [PubMed]
- Cremers, F.P.M.; Lee, W.; Collin, R.W.J.; Allikmets, R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res. 2020, 79, 100861. [Google Scholar] [CrossRef] [PubMed]
- Noupuu, K.; Lee, W.; Zernant, J.; Greenstein, V.C.; Tsang, S.; Allikmets, R. Recessive Stargardt disease phenocopying hydroxychloroquine retinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 2016, 254, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Shroyer, N.F.; Lewis, R.A.; Lupski, J.R. Analysis of the ABCR (ABCA4) gene in 4-aminoquinoline retinopathy: Is retinal toxicity by chloroquine and hydroxychloroquine related to Stargardt disease? Am. J. Ophthalmol. 2001, 131, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, K. InterVar: Clinical interpretation of genetic variants by ACMG-AMP 2015 guideline. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Fokkema, I.F.; Taschner, P.E.; Schaafsma, G.C.; Celli, J.; Laros, J.F.; den Dunnen, J.T. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Jian, X.; Boerwinkle, E.; Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014, 42, 13534–13544. [Google Scholar] [CrossRef]
- Shihab, H.A.; Rogers, M.F.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.M.; Gaunt, T.R.; Campbell, C. An Integrative Approach to Predicting the Functional Consequences of Non-coding and Coding Sequence Variation. Bioinformatics 2015, 31, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Ellard, S.; Baple, E.L.; Berry, I.; Forrester, N.; Turnbull, C.; Owens, M.; Eccles, D.M.; Abbs, S.; Scott, R.; Deans, Z.; et al. ACGS Best Practice Guidelines for Variant Classification 2020: Association for Clinical Genetics Science (ACGS). 2020. Available online: https://www.acgs.uk.com/quality/best-practice-guidelines/#VariantGuidelines (accessed on 4 July 2023).
- Kohno, H.; Maeda, T.; Perusek, L.; Pearlman, E.; Maeda, A. CCL3 production by microglial cells modulates disease severity in murine models of retinal degeneration. J. Immunol. 2014, 192, 3816–3827. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikov, A.V.; Maeda, A.; Tang, P.H.; Imanishi, Y.; Palczewski, K.; Kefalov, V.J. Retinol dehydrogenase 8 and ATP-binding cassette transporter 4 modulate dark adaptation of M-cones in mammalian retina. J. Physiol. 2015, 593, 4923–4941. [Google Scholar] [CrossRef]
- Rattner, A.; Smallwood, P.M.; Nathans, J. Identification and characterization of all-trans-retinol dehydrogenase from photoreceptor outer segments, the visual cycle enzyme that reduces all-trans-retinal to all-trans-retinol. J. Biol. Chem. 2000, 275, 11034–11043. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Maeda, T.; Imanishi, Y.; Kuksa, V.; Alekseev, A.; Bronson, J.D.; Zhang, H.; Zhu, L.; Sun, W.; Saperstein, D.A.; et al. Role of photoreceptor-specific retinol dehydrogenase in the retinoid cycle in vivo. J. Biol. Chem. 2005, 280, 18822–18832. [Google Scholar] [CrossRef]
- Maeda, A.; Maeda, T.; Golczak, M.; Palczewski, K. Retinopathy in mice induced by disrupted all-trans-retinal clearance. J. Biol. Chem. 2008, 283, 26684–26693. [Google Scholar] [CrossRef]
- Chen, C.; Thompson, D.A.; Koutalos, Y. Reduction of all-trans-retinal in vertebrate rod photoreceptors requires the combined action of RDH8 and RDH12. J. Biol. Chem. 2012, 287, 24662–24670. [Google Scholar] [CrossRef]
- Pan, C.; Banerjee, K.; Lehmann, G.L.; Almeida, D.; Hajjar, K.A.; Benedicto, I.; Jiang, Z.; Radu, R.A.; Thompson, D.H.; Rodriguez-Boulan, E.; et al. Lipofuscin causes atypical necroptosis through lysosomal membrane permeabilization. Proc. Natl. Acad. Sci USA 2021, 118, e2100122118. [Google Scholar] [CrossRef] [PubMed]
- Hädicke, A.; Coutinho, A.; Roy, S.; Otis, F.; Lhor, M.; Cantin, L.; Prieto, M.; Voyer, N.; Salesse, C. Membrane binding properties of the C-terminal segment of retinol dehydrogenase 8. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183605. [Google Scholar] [CrossRef] [PubMed]
- von Lintig, J.; Kiser, P.D.; Golczak, M.; Palczewski, K. The biochemical and structural basis for trans-to-cis isomerization of retinoids in the chemistry of vision. Trends Biochem. Sci. 2010, 35, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Kiser, P.D.; Golczak, M.; Maeda, A.; Palczewski, K. Key enzymes of the retinoid (visual) cycle in vertebrate retina. Biochim. Biophys. Acta 2012, 1821, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.J.; Crouch, R.K.; Wiggert, B.; Cornwall, M.C.; Chader, G.J. Retinoid requirements for recovery of sensitivity after visual-pigment bleaching in isolated photoreceptors. Proc. Natl. Acad. Sci. USA 1989, 86, 9606–9610. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Kefalov, V.J. An alternative pathway mediates the mouse and human cone visual cycle. Curr. Biol. 2009, 19, 1665–1669. [Google Scholar] [CrossRef] [PubMed]
- Boyer, N.P.; Thompson, D.A.; Koutalos, Y. Relative Contributions of All-Trans and 11-Cis Retinal to Formation of Lipofuscin and A2E Accumulating in Mouse Retinal Pigment Epithelium. Investig. Ophthalmol. Vis. Sci. 2021, 62, 1. [Google Scholar] [CrossRef] [PubMed]
- Quazi, F.; Molday, R.S. ATP-binding cassette transporter ABCA4 and chemical isomerization protect photoreceptor cells from the toxic accumulation of excess 11-cis-retinal. Proc Natl Acad Sci USA. 2014, 111, 5024–5029. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Cai, B.; Jang, Y.P.; Zhou, J.; Nakanishi, K. A2E, a Fluorophore of RPE Lipofuscin, Can Destabilize Membrane. Adv. Exp. Med. Biol. 2006, 572, 63–68. [Google Scholar]
- Shiose, S.; Chen, Y.; Okano, K.; Roy, S.; Kohno, H.; Tang, J.; Pearlman, E.; Maeda, T.; Palczewski, K.; Maeda, A. Toll-like receptor 3 is required for development of retinopathy caused by impaired all-trans-retinal clearance in mice. J. Biol. Chem. 2011, 286, 15543–15555. [Google Scholar] [CrossRef]
- Kohno, H.; Chen, Y.; Kevany, B.M.; Pearlman, E.; Miyagi, M.; Maeda, T.; Palczewski, K.; Maeda, A. Photoreceptor proteins initiate microglial activation via Toll-like receptor 4 in retinal degeneration mediated by all-transretinal. J. Biol. Chem. 2013, 288, 15326–15341. [Google Scholar] [CrossRef]
- Fabrizio, C.; Termine, A.; Caputo, V.; Megalizzi, D.; Zampatti, S.; Falsini, B.; Cusumano, A.; Eandi, C.M.; Ricci, F.; Giardina, E.; et al. WARE: Wet AMD Risk-Evaluation Tool as a Clinical Decision-Support System Integrating Genetic and Non-Genetic Factors. J. Pers. Med. 2022, 12, 1034. [Google Scholar] [CrossRef]
- Reiley, J.; Botas, P.; Miller, C.E.; Zhao, J.; Malone Jenkins, S.; Best, H.; Grubb, P.H.; Mao, R.; Isla, J.; Brunelli, L. Open-Source Artificial Intelligence System Supports Diagnosis of Mendelian Diseases in Acutely Ill Infants. Children 2023, 10, 991. [Google Scholar] [CrossRef]
- Cadamuro, J.; Cabitza, F.; Debeljak, Z.; De Bruyne, S.; Frans, G.; Perez, S.M.; Ozdemir, H.; Tolios, A.; Carobene, A.; Padoan, A. Potentials and pitfalls of ChatGPT and natural-language artificial intelligence models for the understanding of laboratory medicine test results. An assessment by the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) Working Group on Artificial Intelligence (WG-AI). Clin. Chem. Lab. Med. 2023, 61, 1158–1166. [Google Scholar] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zampatti, S.; Peconi, C.; Calvino, G.; Ferese, R.; Gambardella, S.; Cascella, R.; Sebastiani, J.; Falsini, B.; Cusumano, A.; Giardina, E. A Splicing Variant in RDH8 Is Associated with Autosomal Recessive Stargardt Macular Dystrophy. Genes 2023, 14, 1659. https://doi.org/10.3390/genes14081659
Zampatti S, Peconi C, Calvino G, Ferese R, Gambardella S, Cascella R, Sebastiani J, Falsini B, Cusumano A, Giardina E. A Splicing Variant in RDH8 Is Associated with Autosomal Recessive Stargardt Macular Dystrophy. Genes. 2023; 14(8):1659. https://doi.org/10.3390/genes14081659
Chicago/Turabian StyleZampatti, Stefania, Cristina Peconi, Giulia Calvino, Rosangela Ferese, Stefano Gambardella, Raffaella Cascella, Jacopo Sebastiani, Benedetto Falsini, Andrea Cusumano, and Emiliano Giardina. 2023. "A Splicing Variant in RDH8 Is Associated with Autosomal Recessive Stargardt Macular Dystrophy" Genes 14, no. 8: 1659. https://doi.org/10.3390/genes14081659
APA StyleZampatti, S., Peconi, C., Calvino, G., Ferese, R., Gambardella, S., Cascella, R., Sebastiani, J., Falsini, B., Cusumano, A., & Giardina, E. (2023). A Splicing Variant in RDH8 Is Associated with Autosomal Recessive Stargardt Macular Dystrophy. Genes, 14(8), 1659. https://doi.org/10.3390/genes14081659