
Advancing Epidemiology and Genetic Approaches for the Treatment of Spinal and Bulbar Muscular Atrophy: Focus on Prevalence in the Indigenous Population of Western Canada



{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Clinical Features and Prevalence in the Indigenous Population of Western Canada
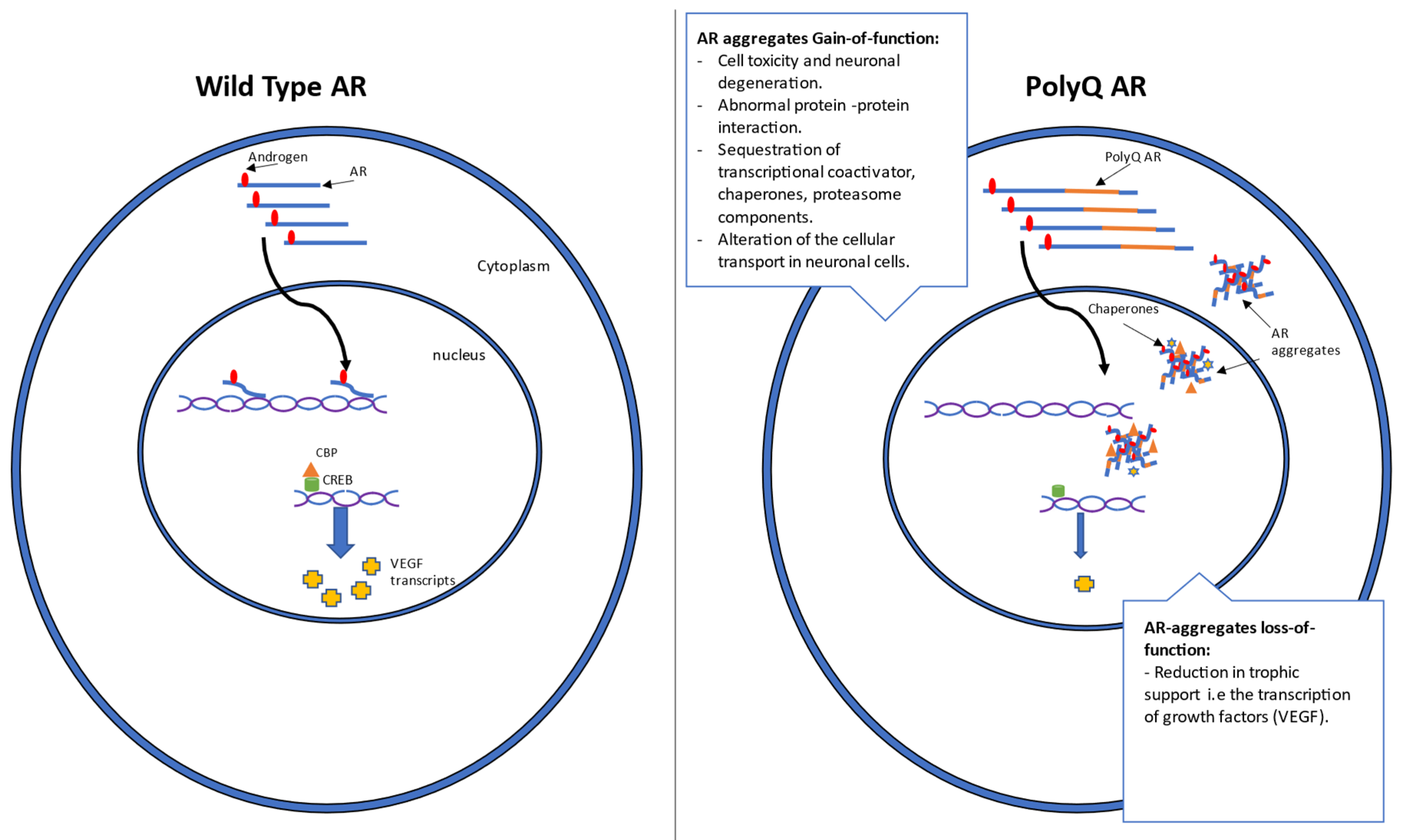
1.2. Mechanism and Genetics
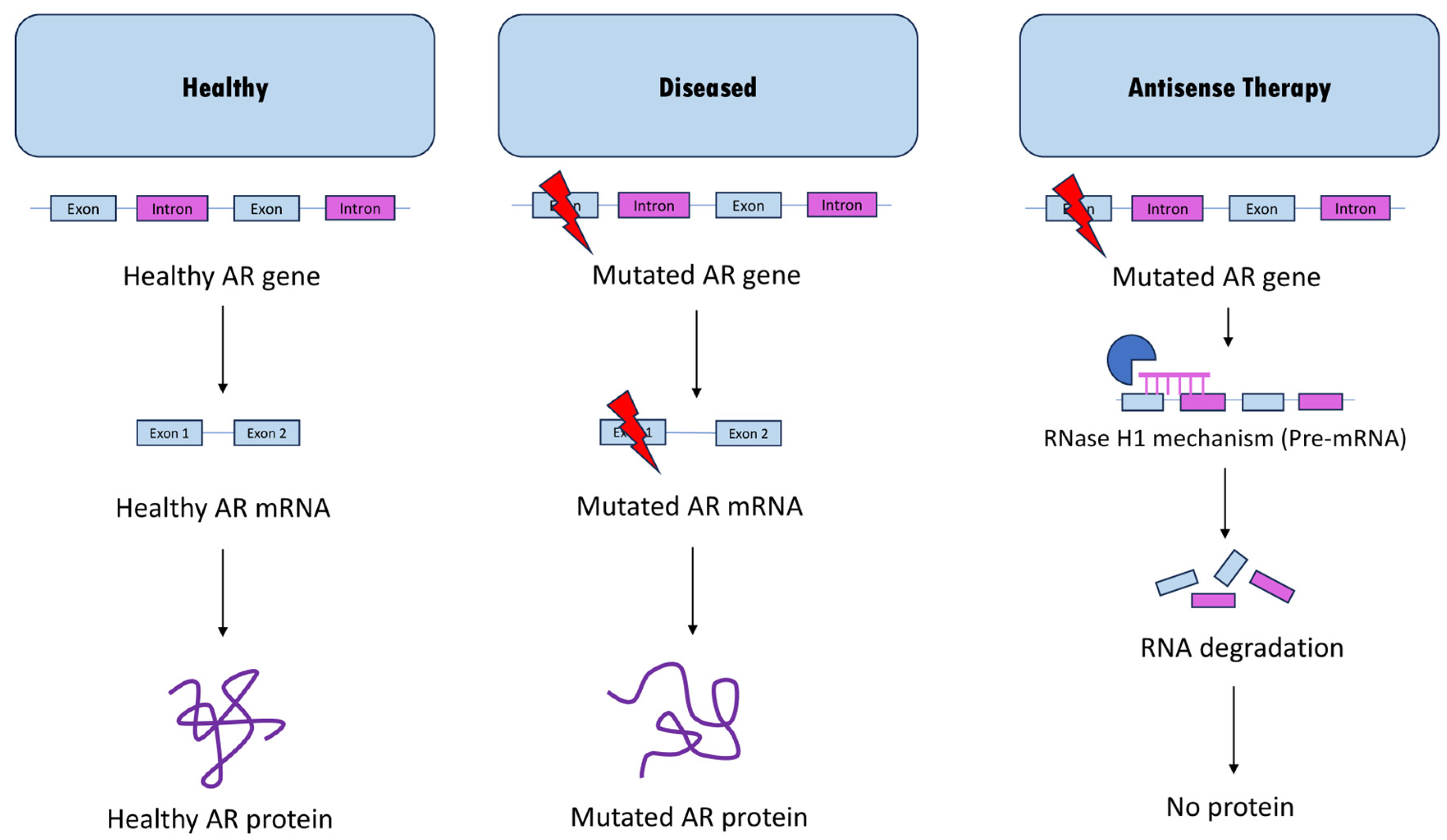
2. Current Antisense Approaches for SBMA
2.1. ASO-Mediated Androgen Receptor Knockdown
2.2. Role of AR45 Isoform in Regulating Androgen Receptor Activity: Potential Therapeutic Implications for SBMA
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kennedy, W.R.; Alter, M.; Sung, J.H. Progressive Proximal Spinal and Bulbar Muscular Atrophy of Late Onset. Neurology 1968, 18, 671. [Google Scholar] [CrossRef]
- Banno, H.; Katsuno, M.; Suzuki, K.; Tanaka, F.; Sobue, G. Pathogenesis and Molecular Targeted Therapy of Spinal and Bulbar Muscular Atrophy (SBMA). Cell Tissue Res. 2012, 349, 313–320. [Google Scholar] [CrossRef]
- Atsuta, N.; Watanabe, H.; Ito, M.; Banno, H.; Suzuki, K.; Katsuno, M.; Tanaka, F.; Tamakoshi, A.; Sobue, G. Natural History of Spinal and Bulbar Muscular Atrophy (SBMA): A Study of 223 Japanese Patients. Brain 2006, 129, 1446–1455. [Google Scholar] [CrossRef] [PubMed]
- Malena, A.; Pennuto, M.; Tezze, C.; Querin, G.; D’Ascenzo, C.; Silani, V.; Cenacchi, G.; Scaramozza, A.; Romito, S.; Morandi, L.; et al. Androgen-Dependent Impairment of Myogenesis in Spinal and Bulbar Muscular Atrophy. Acta Neuropathol. 2013, 126, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Sorarù, G.; D’Ascenzo, C.; Polo, A.; Palmieri, A.; Baggio, L.; Vergani, L.; Gellera, C.; Moretto, G.; Pegoraro, E.; Angelini, C. Spinal and Bulbar Muscular Atrophy: Skeletal Muscle Pathology in Male Patients and Heterozygous Females. J. Neurol. Sci. 2008, 264, 100–105. [Google Scholar] [CrossRef]
- Giorgetti, E.; Lieberman, A.P. Polyglutamine Androgen Receptor-Mediated Neuromuscular Disease. Cell Mol. Life Sci. 2016, 73, 3991. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Yu, Z.; Murray, S.; Peralta, R.; Low, A.; Guo, S.; Yu, X.X.; Cortes, C.J.; Bennett, C.F.; Monia, B.P.; et al. Peripheral Androgen Receptor Gene Suppression Rescues Disease in Mouse Models of Spinal and Bulbar Muscular Atrophy. Cell Rep. 2014, 7, 774–784. [Google Scholar] [CrossRef]
- Dejager, S.; Bry-Gauillard, H.; Bruckert, E.; Eymard, B.; Salachas, F.; LeGuern, E.; Tardieu, S.; Chadarevian, R.; Giral, P.; Turpin, G. A Comprehensive Endocrine Description of Kennedy’s Disease Revealing Androgen Insensitivity Linked to CAG Repeat Length. J. Clin. Endocrinol. Metab. 2002, 87, 3893–3901. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rosenbohm, A.; Hirsch, S.; Volk, A.E.; Grehl, T.; Grosskreutz, J.; Hanisch, F.; Herrmann, A.; Kollewe, K.; Kress, W.; Meyer, T.; et al. The Metabolic and Endocrine Characteristics in Spinal and Bulbar Muscular Atrophy. J. Neurol. 2018, 265, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Querin, G.; Bertolin, C.; Da Re, E.; Volpe, M.; Zara, G.; Pegoraro, E.; Caretta, N.; Foresta, C.; Silvano, M.; Corrado, D.; et al. Non-Neural Phenotype of Spinal and Bulbar Muscular Atrophy: Results from a Large Cohort of Italian Patients. J. Neurol. Neurosurg. Psychiatry 2016, 87, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Francini-Pesenti, F.; Querin, G.; Martini, C.; Mareso, S.; Sacerdoti, D. Prevalence of Metabolic Syndrome and Non-Alcoholic Fatty Liver Disease in a Cohort of Italian Patients with Spinal-Bulbar Muscular Atrophy. Acta Myol. 2018, 37, 204. [Google Scholar] [PubMed]
- Tanaka, F.; Katsuno, M.; Banno, H.; Suzuki, K.; Adachi, H.; Sobue, G. Current Status of Treatment of Spinal and Bulbar Muscular Atrophy. Neural Plast. 2012, 2012, 369284. [Google Scholar] [CrossRef]
- Pradat, P.F.; Bernard, E.; Corcia, P.; Couratier, P.; Jublanc, C.; Querin, G.; Morélot Panzini, C.; Salachas, F.; Vial, C.; Wahbi, K.; et al. The French National Protocol for Kennedy’s Disease (SBMA): Consensus Diagnostic and Management Recommendations. Orphanet J. Rare Dis. 2020, 15, 90. [Google Scholar] [CrossRef]
- Guidetti, D.; Sabadini, R.; Ferlini, A.; Torrente, I. Epidemiological Survey of X-Linked Bulbar and Spinal Muscular Atrophy, or Kennedy Disease, in the Province of Reggio Emilia, Italy. Eur. J. Epidemiol. 2001, 17, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.; Udd, B.; Juvonen, V.; Andersen, P.M.; Cederquist, K.; Davis, M.; Gellera, C.; Kölmel, C.; Ronnevi, L.O.; Sperfeld, A.D.; et al. Multiple Founder Effects in Spinal and Bulbar Muscular Atrophy (SBMA, Kennedy Disease) around the World. Eur. J. Hum. Genet. 2001, 9, 431–436. [Google Scholar] [CrossRef]
- Leckie, J.N.; Joel, M.M.; Martens, K.; King, A.; King, M.; Korngut, L.W.; de Koning, A.P.J.; Pfeffer, G.; Schellenberg, K.L. Highly Elevated Prevalence of Spinobulbar Muscular Atrophy in Indigenous Communities in Canada Due to a Founder Effect. Neurol. Genet. 2021, 7, e607. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.; Udd, B.; Juvonen, V.; Andersen, P.M.; Cederquist, K.; Ronnevi, L.O.; Sistonen, P.; Sörensen, S.A.; Tranebjærg, L.; Wallgren-Pettersson, C.; et al. Founder Effect in Spinal and Bulbar Muscular Atrophy (SBMA) in Scandinavia. Eur. J. Hum. Genet. 2000, 8, 631–636. [Google Scholar] [CrossRef]
- Tanaka, F.; Doyu, M.; Ito, Y.; Matsumoto, M.; Mitsuma, T.; Abe, K.; Aoki, M.; Itoyama, Y.; Fischbeck, K.H.; Sobue, G. Founder Effect in Spinal and Bulbar Muscular Atrophy (SBMA). Hum. Mol. Genet. 1996, 5, 1253–1257. [Google Scholar] [CrossRef]
- King, M.; Smith, A.; Gracey, M. Indigenous Health Part 2: The Underlying Causes of the Health Gap. Lancet 2009, 374, 76–85. [Google Scholar] [CrossRef]
- Martin, D.; Miller, A.P.; Quesnel-Vallée, A.; Caron, N.R.; Vissandjée, B.; Marchildon, G.P. Canada’s Universal Health-Care System: Achieving Its Potential. Lancet 2018, 391, 1718–1735. [Google Scholar] [CrossRef]
- Caron, N.R.; Chongo, M.; Hudson, M.; Arbour, L.; Wasserman, W.W.; Robertson, S.; Correard, S.; Wilcox, P. Indigenous Genomic Databases: Pragmatic Considerations and Cultural Contexts. Front. Public Health 2020, 8, 529095. [Google Scholar] [CrossRef] [PubMed]
- Yong, E.L.; Loy, C.J.; Sim, K.S. Androgen Receptor Gene and Male Infertility. Hum. Reprod. Update 2003, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.Z.; Wardell, S.E.; Burnstein, K.L.; Defranco, D.; Fuller, P.J.; Giguere, V.; Hochberg, R.B.; Mckay, L.; Renoir, J.M.; Weigel, N.L.; et al. International Union of Pharmacology. LXV. The Pharmacology and Classification of the Nuclear Receptor Superfamily: Glucocorticoid, Mineralocorticoid, Progesterone, and Androgen Receptors. Pharmacol. Rev. 2006, 58, 782–797. [Google Scholar] [CrossRef] [PubMed]
- Palazzolo, I.; Gliozzi, A.; Rusmini, P.; Sau, D.; Crippa, V.; Simonini, F.; Onesto, E.; Bolzoni, E.; Poletti, A. The Role of the Polyglutamine Tract in Androgen Receptor. J. Steroid Biochem. Mol. Biol. 2008, 108, 245–253. [Google Scholar] [CrossRef]
- Brown, C.J.; Goss, S.J.; Lubahn, D.B.; Joseph, D.R.; Wilson, E.M.; French, F.S.; Willard, H.F. Androgen Receptor Locus on the Human X Chromosome: Regional Localization to Xq11-12 and Description of a DNA Polymorphism. Am. J. Hum. Genet. 1989, 44, 264. [Google Scholar]
- Lubahn, D.B.; Joseph, D.R.; Sullivan, P.M.; Willard, H.F.; French, F.S.; Wilson, E.M. Cloning of Human Androgen Receptor Complementary DNA and Localization to the X Chromosome. Science 1988, 240, 327–330. [Google Scholar] [CrossRef]
- Chang, C.; Kokontis, J.; Liao, S. Molecular Cloning of Human and Rat Complementary DNA Encoding Androgen Receptors. Science 1988, 240, 324–326. [Google Scholar] [CrossRef]
- van Laar, J.H.; de Vries, J.B.; Voorhorst-Ogink, M.M.; Brinkmann, A.O. The Human Androgen Receptor Is a 110 KDa Protein. Mol. Cell Endocrinol. 1989, 63, 39–44. [Google Scholar] [CrossRef]
- Kuiper, G.G.J.M.; Faber, P.W.; Van Rooij, H.C.J.; Van der Korput, J.A.G.M.; Ris-Stalpers, C.; Klaassen, P.; Trapman, J.; Brinkmann, A.O. Structural organization of the human androgen receptor gene. J. Mol. Endocrinol. 1989, 2, R1–R4. [Google Scholar] [CrossRef]
- Tsai, M.J.; O’Malley, B.W. Molecular Mechanisms of Action of Steroid/Thyroid Receptor Superfamily Members. Annu. Rev. Biochem. 1994, 63, 451–486. [Google Scholar] [CrossRef]
- Brinkmann, A.O. Molecular Basis of Androgen Insensitivity. Mol. Cell Endocrinol. 2001, 179, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Fischbeck, K.H.; Lieberman, A.; Bailey, C.K.; Abel, A.; Merry, D.E. Androgen Receptor Mutation in Kennedy’s Disease. Philos. Trans. R. Soc. B: Biol. Sci. 1999, 354, 1075. [Google Scholar] [CrossRef][Green Version]
- Fratta, P.; Collins, T.; Pemble, S.; Nethisinghe, S.; Devoy, A.; Giunti, P.; Sweeney, M.G.; Hanna, M.G.; Fisher, E.M.C. Sequencing Analysis of the Spinal Bulbar Muscular Atrophy CAG Expansion Reveals Absence of Repeat Interruptions. Neurobiol. Aging 2014, 35, 443.e1. [Google Scholar] [CrossRef] [PubMed]
- Stoyas, C.A.; La Spada, A.R. The CAG–Polyglutamine Repeat Diseases: A Clinical, Molecular, Genetic, and Pathophysiologic Nosology. Handb. Clin. Neurol. 2018, 147, 143–170. [Google Scholar] [CrossRef]
- Gelmann, E.P. Molecular Biology of the Androgen Receptor. J. Clin. Oncol. 2002, 20, 3001–3015. [Google Scholar] [CrossRef]
- Ing, N.H.; Beekman, J.M.; Tsai, S.Y.; Tsai, M.-J.; O’malleyl, B.W.; Sagami, I.; Tsai, S.Y.; Wang, H.; Tsii, M.-J.; Malley, O.; et al. Members of the Steroid Hormone Receptor Superfamily Interact with TFIIB (S300-11). J. Biol. Chem. 1992, 267, 17617–17623. [Google Scholar] [CrossRef]
- Baniahmad, A.; Ha, I.; Reinberg, D.; Tsai, S.; Tsai, M.J.; O’Malley, B.W. Interaction of Human Thyroid Hormone Receptor Beta with Transcription Factor TFIIB May Mediate Target Gene Derepression and Activation by Thyroid Hormone. Proc. Natl. Acad. Sci. USA 1993, 90, 8832–8836. [Google Scholar] [CrossRef] [PubMed]
- Brou, C.; Chaudhary, S.; Davidson, I.; Lutz, Y.; Wu, J.; Egly, J.M.; Tora, L.; Chambon, P. Distinct TFIID Complexes Mediate the Effect of Different Transcriptional Activators. EMBO J. 1993, 12, 489–499. [Google Scholar] [CrossRef]
- Brou, C.; Wu, J.; Ali, S.; Scheer, E.; Lang, C.; Davidson, I.; Chambon, P.; Tora, L. Different TBP-Associated Factors Are Required for Mediating the Stimulation of Transcription in Vitro by the Acidic Transactivator GAL-VP16 and the Two Nonacidic Activation Functions of the Estrogen Receptor. Nucleic Acids Res. 1993, 21, 5. [Google Scholar] [CrossRef]
- Mcewan, I.J.; Gustafsson, J.Å. Interaction of the Human Androgen Receptor Transactivation Function with the General Transcription Factor TFIIF. Proc. Natl. Acad. Sci. USA 1997, 94, 8485. [Google Scholar] [CrossRef]
- Schulman, I.G.; Chakravarti, D.; Juguilon, H.; Romo, A.; Evans, R.M. Interactions between the Retinoid X Receptor and a Conserved Region of the TATA-Binding Protein Mediate Hormone-Dependent Transactivation. Proc. Natl. Acad. Sci. USA 1995, 92, 8288. [Google Scholar] [CrossRef]
- Thomas, P.S.; Fraley, G.S.; Damien, V.; Woodke, L.B.; Zapata, F.; Sopher, B.L.; Plymate, S.R.; La Spada, A.R. Loss of Endogenous Androgen Receptor Protein Accelerates Motor Neuron Degeneration and Accentuates Androgen Insensitivity in a Mouse Model of X-Linked Spinal and Bulbar Muscular Atrophy. Hum. Mol. Genet. 2006, 15, 2225–2238. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, H.Y.; Orr, H.T. Glutamine Repeats and Neurodegeneration. Annu. Rev. Neurosci. 2000, 23, 217–247. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A. When More Is Less: Pathogenesis of Glutamine Repeat Neurodegenerative Diseases. Neuron 1995, 15, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Craig, T.J.; Henley, J.M. Fighting Polyglutamine Disease by Wrestling with SUMO. J. Clin. Investig. 2015, 125, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Nakagomi, Y.; Kobayashi, Y.; Merry, D.E.; Tanaka, F.; Doyu, M.; Mitsuma, T.; Hashizume, Y.; Fischbeck, K.H.; Sobue, G. Nonneural Nuclear Inclusions of Androgen Receptor Protein in Spinal and Bulbar Muscular Atrophy. Am. J. Pathol. 1998, 153, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Arnold, F.J.; Merry, D.E. Molecular Mechanisms and Therapeutics for SBMA/Kennedy’s Disease. Neurotherapeutics 2019, 16, 928–947. [Google Scholar] [CrossRef]
- Davies, P.; Watt, K.; Kelly, S.M.; Clark, C.; Price, N.C.; McEwan, I.J. Consequences of Poly-Glutamine Repeat Length for the Conformation and Folding of the Androgen Receptor Amino-Terminal Domain. J. Mol. Endocrinol. 2008, 41, 301–314. [Google Scholar] [CrossRef]
- Escobedo, A.; Topal, B.; Kunze, M.B.A.; Aranda, J.; Chiesa, G.; Mungianu, D.; Bernardo-Seisdedos, G.; Eftekharzadeh, B.; Gairí, M.; Pierattelli, R.; et al. Side Chain to Main Chain Hydrogen Bonds Stabilize a Polyglutamine Helix in a Transcription Factor. Nat. Commun. 2019, 10, 2034. [Google Scholar] [CrossRef]
- Perutz, M.F.; Johnson, T.; Suzuki, M.; Finch, J.T. Glutamine Repeats as Polar Zippers: Their Possible Role in Inherited Neurodegenerative Diseases. Proc. Natl. Acad. Sci. USA 1994, 91, 5355. [Google Scholar] [CrossRef]
- Green, H. Human Genetic Diseases Due to Codon Reiteration: Relationship to an Evolutionary Mechanism. Cell 1993, 74, 955–956. [Google Scholar] [CrossRef]
- Morfini, G.; Pigino, G.; Szebenyi, G.; You, Y.; Pollema, S.; Brady, S.T. JNK Mediates Pathogenic Effects of Polyglutamine-Expanded Androgen Receptor on Fast Axonal Transport. Nat. Neurosci. 2006, 9, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Rgyi Szebenyi, G.; Morfini, G.A.; Babcock, A.; Gould, M.; Selkoe, K.; Stenoien, D.L.; Young, M.; Faber, P.W.; Macdonald, M.E.; Mcphaul, M.J.; et al. Neuropathogenic Forms of Huntingtin and Androgen Receptor Inhibit Fast Axonal Transport. Neuron 2003, 40, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Piccioni, F.; Pinton, P.; Simeoni, S.; Pozzi, P.; Fascio, U.; Vismara, G.; Martini, L.; Rizzuto, R.; Poletti, A. Androgen Receptor with Elongated Polyglutamine Tract Forms Aggregates That Alter Axonal Trafficking and Mitochondrial Distribution in Motor Neuronal Processes. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1418–1420. [Google Scholar] [CrossRef]
- Sopher, B.L.; Thomas, P.S.; Lafevre-Bernt, M.A.; Holm, I.E.; Wilke, S.A.; Ware, C.B.; Jin, L.W.; Libby, R.T.; Ellerby, L.M.; La Spada, A.R. Androgen Receptor YAC Transgenic Mice Recapitulate SBMA Motor Neuronopathy and Implicate VEGF164 in the Motor Neuron Degeneration. Neuron 2004, 41, 687–699. [Google Scholar] [CrossRef]
- Crooke, S.T.; Baker, B.F.; Crooke, R.M.; Liang, X. hai Antisense Technology: An Overview and Prospectus. Nat. Rev. Drug Discov. 2021, 20, 427–453. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous Sarcoma Virus Replication and Cell Transformation by a Specific Oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280. [Google Scholar] [CrossRef]
- Wu, H.; Lima, W.F.; Zhang, H.; Fan, A.; Sun, H.; Crooke, S.T. Determination of the Role of the Human RNase H1 in the Pharmacology of DNA-like Antisense Drugs. J. Biol. Chem. 2004, 279, 17181–17189. [Google Scholar] [CrossRef]
- Gagliardi, M.; Ashizawa, A.T. The Challenges and Strategies of Antisense Oligonucleotide Drug Delivery. Biomedicines 2021, 9, 433. [Google Scholar] [CrossRef]
- Havens, M.A.; Hastings, M.L. Splice-Switching Antisense Oligonucleotides as Therapeutic Drugs. Nucleic Acids Res. 2016, 44, 6549. [Google Scholar] [CrossRef]
- Sahashi, K.; Katsuno, M.; Hung, G.; Adachi, H.; Kondo, N.; Nakatsuji, H.; Tohnai, G.; Iida, M.; Bennett, F.F.; Sobue, G. Silencing Neuronal Mutant Androgen Receptor in a Mouse Model of Spinal and Bulbar Muscular Atrophy. Hum. Mol. Genet. 2015, 24, 5985–5994. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.M.; Pepers, B.A.; van Deutekom, J.C.T.; Mulders, S.A.M.; den Dunnen, J.T.; Aartsma-Rus, A.; van Ommen, G.J.B.; van Roon-Mom, W.M.C. Targeting Several CAG Expansion Diseases by a Single Antisense Oligonucleotide. PLoS ONE 2011, 6, e24308. [Google Scholar] [CrossRef] [PubMed]
- Hamy, F.; Brondani, V.; Spoerri, R.; Rigo, S.; Stamm, C.; Klimkait, T. Specific Block of Androgen Receptor Activity by Antisense Oligonucleotides. Prostate Cancer Prostatic Dis. 2003, 6, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Loriot, Y.; Beraldi, E.; Zhang, F.; Wyatt, A.W.; Al Nakouzi, N.; Mo, F.; Zhou, T.; Kim, Y.; Monia, B.P.; et al. Generation 2.5 Antisense Oligonucleotides Targeting the Androgen Receptor and Its Splice Variants Suppress Enzalutamide-Resistant Prostate Cancer Cell Growth. Clin. Cancer Res. 2015, 21, 1675–1687. [Google Scholar] [CrossRef]
- Wang, Y.; Pang, J.; Wang, Q.; Yan, L.; Wang, L.; Xing, Z.; Wang, C.; Zhang, J.; Dong, L.; Wang, Y.; et al. Delivering Antisense Oligonucleotides across the Blood-Brain Barrier by Tumor Cell-Derived Small Apoptotic Bodies. Adv. Sci. 2021, 8, 2004929. [Google Scholar] [CrossRef]
- Sun, Y.; Kong, J.; Ge, X.; Mao, M.; Yu, H.; Wang, Y. An Antisense Oligonucleotide-Loaded Blood-Brain Barrier Penetrable Nanoparticle Mediating Recruitment of Endogenous Neural Stem Cells for the Treatment of Parkinson’s Disease. ACS Nano 2023, 17, 4414–4432. [Google Scholar] [CrossRef]
- Min, H.S.; Kim, H.J.; Naito, M.; Ogura, S.; Toh, K.; Hayashi, K.; Kim, B.S.; Fukushima, S.; Anraku, Y.; Miyata, K.; et al. Systemic Brain Delivery of Antisense Oligonucleotides across the Blood–Brain Barrier with a Glucose-Coated Polymeric Nanocarrier. Angew. Chem. Int. Ed. 2020, 59, 8173–8180. [Google Scholar] [CrossRef]
- Ahrens-Fath, I.; Politz, O.; Geserick, C.; Haendler, B. Androgen Receptor Function Is Modulated by the Tissue-Specific AR45 Variant. FEBS J. 2005, 272, 74–84. [Google Scholar] [CrossRef]
- Wilson, C.M.; Mcphaul, M.J. A and B Forms of the Androgen Receptor Are Present in human Genital Skin Fibroblasts. Proc. Natl. Acad. Sci. USA 1994, 91, 1234. [Google Scholar] [CrossRef]
- Dehm, S.M.; Tindall, D.J. Alternatively Spliced Androgen Receptor Variants. Endocr. Relat. Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen Receptor Splice Variant-7 Expression Emerges with Castration Resistance in Prostate Cancer. J. Clin. Investig. 2018, 129, 192. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med. 2014, 371, 1028. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Karsh, L.I.; Nissenblatt, M.J.; Canfield, S.E. Androgen Receptor Splice Variant, AR-V7, as a Biomarker of Resistance to Androgen Axis-Targeted Therapies in Advanced Prostate Cancer. Clin. Genitourin. Cancer 2020, 18, 1–10. [Google Scholar] [CrossRef]
- Lim, W.F.; Forouhan, M.; Roberts, T.C.; Dabney, J.; Ellerington, R.; Speciale, A.A.; Manzano, R.; Lieto, M.; Sangha, G.; Banerjee, S.; et al. Gene Therapy with AR Isoform 2 Rescues Spinal and Bulbar Muscular Atrophy Phenotype by Modulating AR Transcriptional Activity. Sci. Adv. 2021, 7, eabi6896. [Google Scholar] [CrossRef] [PubMed]
- Heidersbach, A.J.; Dorighi, K.M.; Gomez, J.A.; Jacobi, A.M.; Haley, B. A Versatile, High-Efficiency Platform for CRISPR-Based Gene Activation. Nat. Commun. 2023, 14, 1–10. [Google Scholar] [CrossRef]
- Di Maria, V.; Moindrot, M.; Ryde, M.; Bono, A.; Quintino, L.; Ledri, M. Development and Validation of CRISPR Activator Systems for Overexpression of CB1 Receptors in Neurons. Front. Mol. Neurosci. 2020, 13, 526700. [Google Scholar] [CrossRef]
- Kwok, A.; Raulf, N.; Habib, N. Developing Small Activating RNA as a Therapeutic: Current Challenges and Promises. Ther. Deliv. 2019, 10, 151–164. [Google Scholar] [CrossRef]
- Ghanbarian, H.; Aghamiri, S.; Eftekhary, M.; Wagner, N.; Wagner, K.D. Small Activating RNAs: Towards the Development of New Therapeutic Agents and Clinical Treatments. Cells 2021, 10, 591. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Woo, S.; Melo, D.; Huang, Y.; Dzierlega, K.; Shah, M.N.A.; Aslesh, T.; Roshmi, R.R.; Echigoya, Y.; Maruyama, R.; et al. Development of DG9 Peptide-Conjugated Single- and Multi-Exon Skipping Therapies for the Treatment of Duchenne Muscular Dystrophy. Proc. Natl. Acad. Sci. USA 2022, 119, e2112546119. [Google Scholar] [CrossRef]
- Moulton, H.M.; Moulton, J.D. Morpholinos and Their Peptide Conjugates: Therapeutic Promise and Challenge for Duchenne Muscular Dystrophy. Biochim. Biophys. Acta (BBA)-Biomembr. 2010, 1798, 2296–2303. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilton-Clark, H.; Al-aghbari, A.; Yang, J.; Yokota, T. Advancing Epidemiology and Genetic Approaches for the Treatment of Spinal and Bulbar Muscular Atrophy: Focus on Prevalence in the Indigenous Population of Western Canada. Genes 2023, 14, 1634. https://doi.org/10.3390/genes14081634
Wilton-Clark H, Al-aghbari A, Yang J, Yokota T. Advancing Epidemiology and Genetic Approaches for the Treatment of Spinal and Bulbar Muscular Atrophy: Focus on Prevalence in the Indigenous Population of Western Canada. Genes. 2023; 14(8):1634. https://doi.org/10.3390/genes14081634
Chicago/Turabian StyleWilton-Clark, Harry, Ammar Al-aghbari, Jessica Yang, and Toshifumi Yokota. 2023. "Advancing Epidemiology and Genetic Approaches for the Treatment of Spinal and Bulbar Muscular Atrophy: Focus on Prevalence in the Indigenous Population of Western Canada" Genes 14, no. 8: 1634. https://doi.org/10.3390/genes14081634
APA StyleWilton-Clark, H., Al-aghbari, A., Yang, J., & Yokota, T. (2023). Advancing Epidemiology and Genetic Approaches for the Treatment of Spinal and Bulbar Muscular Atrophy: Focus on Prevalence in the Indigenous Population of Western Canada. Genes, 14(8), 1634. https://doi.org/10.3390/genes14081634