

Genome-Wide Single-Nucleotide Polymorphism-Based Genomic Diversity and Runs of Homozygosity for Selection Signatures in Equine Breeds
 , , ,
, , ,  , , , , , and
, , , , , and
Abstract
1. Introduction
2. Methodology
2.1. Samples Collection, DNA Extraction and SNP Genotyping
2.2. Data Quality Control and Measurement of Runs of Homozygosity and Their Distribution
2.3. Genomic Inbreeding Coefficients
2.4. Population Diversity Analysis
2.5. Gene Annotation and Functional Enrichment Analysis
2.6. ROH Annotation by Horse QTL Database
2.7. Development of Equine SNP Marker Database
3. Results
3.1. Filtration of Genotypes
3.2. ROH Distribution
3.3. Population Diversity and Structure Analysis
3.4. Gene Annotation and Enrichment Analysis
3.5. QTL Analysis
3.6. Development of Equine SNP Marker Database
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wade, C.M.; Giulotto, E.; Sigurdsson, S.; Zoli, M.; Gnerre, S.; Imsland, F.; Lindblad-Toh, K. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science 2009, 326, 865–867. [Google Scholar] [PubMed]
- Jun, J.; Cho, Y.S.; Hu, H.; Kim, H.M.; Jho, S.; Gadhvi, P.; Bhak, J. Whole genome sequence and analysis of the Marwari horse breed and its genetic origin. BMC Genom. 2014, 15, S4. [Google Scholar]
- Gupta, A.K.; Tandon, S.N.; Pal, Y.; Bhardwaj, A.; Chauhan, M. Phenotypic characterization of Indian equine breeds: A comparative study. Anim. Genet. Resour. 2012, 50, 49–58. [Google Scholar]
- Gupta, A.K.; Chauhan, M.; Bhardwaj, A.; Gupta, N.; Gupta, S.C.; Pal, Y.; Vijh, R.K. Comparative genetic diversity analysis among six Indian breeds and English Thoroughbred horses. Livest. Sci. 2014, 163, 1–11. [Google Scholar]
- Pal, Y.; Legha, R.A.; Bhardwaj, A.; Tripathi, B.N. Status and conservation of equine biodiversity in India. Indian J. Comp. Microbiol. Immunol. Infect. Dis. 2020, 41, 174–184. [Google Scholar]
- Behl, R.; Behl, J.; Gupta, N.; Gupta, S.C. Genetic relationships of five Indian horse breeds using microsatellite markers. Animal 2007, 1, 483–488. [Google Scholar] [PubMed]
- McCue, M.E.; Bannasch, D.L.; Petersen, J.L.; Gurr, J.; Bailey, E.; Binns, M.M.; Mickelson, J.R. A high density SNP array for the domestic horse and extant Perissodactyla: Utility for association mapping, genetic diversity, and phylogeny studies. PLoS Genet. 2012, 8, e1002451. [Google Scholar]
- Schaefer, R.J.; Schubert, M.; Bailey, E.; Bannasch, D.L.; Barrey, E.; Bar-Gal, G.K.; McCue, M.E. Developing a 670k genotyping array to tag~ 2M SNPs across 24 horse breeds. BMC Genom. 2017, 18, 565. [Google Scholar]
- Gupta, A.; Bhardwaj, A.; Sharma, P.; Pal, Y. Mitochondrial DNA-a tool for phylogenetic and biodiversity search in equines. J. Biodivers. Endanger. Species 2015, 1, 1–8. [Google Scholar]
- Rafati, N.; Andersson, L.S.; Mikko, S.; Feng, C.; Raudsepp, T.; Pettersson, J.; Rubin, C.J. Large deletions at the SHOX locus in the pseudoautosomal region are associated with skeletal atavism in Shetland Ponies. G3 Genes Genomes Genet. 2016, 6, 2213–2223. [Google Scholar]
- Grilz-Seger, G.; Neuditschko, M.; Ricard, A.; Velie, B.; Lindgren, G.; Mesarič, M.; Druml, T. Genome-wide homozygosity patterns and evidence for selection in a set of European and near eastern horse breeds. Genes 2019, 10, 491. [Google Scholar] [PubMed]
- Solé, M.; Ablondi, M.; Binzer-Panchal, A.; Velie, B.D.; Hollfelder, N.; Buys, N.; Ducro, B.J.; François, L.; Janssens, S.; Schurink, A.; et al. Inter-and intra-breed genome-wide copy number diversity in a large cohort of European equine breeds. BMC Genom. 2019, 20, 759. [Google Scholar]
- Norton, E.; Schultz, N.; Geor, R.; McFarlane, D.; Mickelson, J.; McCue, M. Genome-wide association analyses of equine metabolic syndrome phenotypes in Welsh ponies and Morgan horses. Genes 2019, 10, 893. [Google Scholar] [PubMed]
- Raudsepp, T.; McCue, M.E.; Das, P.J.; Dobson, L.; Vishnoi, M.; Fritz, K.L.; Schaefer, R.; Rendahl, A.K.; Derr, J.N.; Love, C.C.; et al. Genome-wide association study implicates testis-sperm specific FKBP6 as a susceptibility locus for impaired acrosome reaction in stallions. PLoS Genet. 2012, 8, 1003139. [Google Scholar]
- Fawcett, J.A.; Sato, F.; Sakamoto, T.; Iwasaki, W.M.; Tozaki, T.; Innan, H. Genome-wide SNP analysis of Japanese Thoroughbred racehorses. PLoS ONE 2019, 14, 0218407. [Google Scholar]
- Cosgrove, E.J.; Sadeghi, R.; Schlamp, F.; Holl, H.M.; Moradi-Shahrbabak, M.; Miraei-Ashtiani, S.R.; Brooks, S.A. Genome diversity and the origin of the Arabian horse. Sci. Rep. 2020, 10, 9702. [Google Scholar] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, s13742-015. [Google Scholar]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar]
- Fang, Y.; Hao, X.; Xu, Z.; Sun, H.; Zhao, Q.; Cao, R.; Pan, Y. Genome-wide detection of runs of homozygosity in Laiwu pigs revealed by sequencing data. Front. Genet. 2021, 12, 629966. [Google Scholar]
- Kirin, M.; McQuillan, R.; Franklin, C.S.; Campbell, H.; McKeigue, P.M.; Wilson, J.F. Genomic runs of homozygosity record population history and consanguinity. PLoS ONE 2010, 5, e13996. [Google Scholar]
- Ferenčaković, M.; Hamzić, E.; Gredler, B.; Solberg, T.R.; Klemetsdal, G.; Curik, I.; Sölkner, J. Estimates of autozygosity derived from runs of homozygosity: Empirical evidence from selected cattle populations. J. Anim. Breed. Genet. 2013, 130, 286–293. [Google Scholar]
- Ferenčaković, M.; Sölkner, J.; Curik, I. Estimating autozygosity from high-throughput information: Effects of SNP density and genotyping errors. Genet. Sel. Evol. 2013, 45, 42. [Google Scholar]
- McQuillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef]
- Kaeuffer, R.; Réale, D.; Coltman, D.W.; Pontier, D. Detecting population structure using STRUCTURE software: Effect of background linkage disequilibrium. Heredity 2007, 99, 374–380. [Google Scholar]
- Cingolani, P. Variant annotation and functional prediction: SnpEff. In Variant Calling: Methods and Protocols; Springer: New York, NY, USA, 2022; pp. 289–314. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35 (Suppl. 2), W169–W175. [Google Scholar]
- Willing, E.M.; Dreyer, C.; van Oosterhout, C. Estimates of genetic differentiation measured by FST do not necessarily require large sample sizes when using many SNP markers. PLoS ONE 2012, 7, e42649. [Google Scholar]
- Colli, L.; Milanesi, M.; Vajana, E.; Iamartino, D.; Bomba, L.; Puglisi, F. New insights on water buffalo genomic diversity and postdomestication migration routes from medium density SNP chip data. Front. Genet. 2018, 9, 53. [Google Scholar] [CrossRef]
- Utsunomiya, Y.T.; Milanesi, M.; Fortes, M.R.S.; Porto-Neto, L.R.; Utsunomiya, A.T.H.; Silva, M.V.G.B.; Garcia, J.F.; Ajmone-Marsan, P. Genomic clues of the evolutionary history of Bos indicus cattle. Anim. Genet. 2019, 50, 557–568. [Google Scholar]
- Dzomba, E.F.; Chimonyo, M.; Pierneef, R.; Muchadeyi, F.C. Runs of homozygosity analysis of South African sheep breeds from various production systems investigated using OvineSNP50k data. BMC Genom. 2021, 22, 7. [Google Scholar]
- Xu, Z.; Mei, S.; Zhou, J.; Zhang, Y.; Qiao, M.; Sun, H.; Peng, X. Genome-wide assessment of runs of homozygosity and estimates of genomic inbreeding in a chinese composite pig breed. Front. Genet. 2021, 12, 720081. [Google Scholar]
- Howrigan, D.P.; Simonson, M.A.; Keller, M.C. Detecting autozygosity through runs of homozygosity: A comparison of three autozygosity detection algorithms. BMC Genom. 2011, 12, 460. [Google Scholar]
- Mastrangelo, S.; Tolone, M.; Sardina, M.T.; Sottile, G.; Sutera, A.M.; Di Gerlando, R.; Portolano, B. Genome-wide scan for runs of homozygosity identifies potential candidate genes associated with local adaptation in Valle del Belice sheep. Genet. Sel. Evol. 2017, 49, 84. [Google Scholar]
- Peripolli, E.; Stafuzza, N.B.; Munari, D.P.; Lima, A.L.F.; Irgang, R.; Machado, M.A.; do Carmo Panetto, J.C.; Vieira Ventura, R.; Baldi, F.; da Silva, M.V.G.B. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC Genom. 2018, 19, 34. [Google Scholar]
- Subauste, A.R.; Elliott, B.; Das, A.K.; Burant, C.F. A role for 1-acylglycerol-3-phosphate-O-acyltransferase-1 in myoblast differentiation. Differentiation 2010, 80, 140–146. [Google Scholar]
- Zhu, Q.; Huo, H.; Fu, Q.; Yang, N.; Xue, T.; Zhuang, C.; Liu, X.; Wang, B.; Su, B.; Li, C. Identification and characterization of a C-type lectin in turbot (Scophthalmus maximus) which functioning as a pattern recognition receptor that binds and agglutinates various bacteria. Fish Shellfish Immunol. 2021, 115, 104–111. [Google Scholar]
- Hestand, M.S.; Kalbfleisch, T.S.; Coleman, S.J.; Zeng, Z.; Liu, J.; Orlando, L.; MacLeod, J.N. Annotation of the protein coding regions of the equine genome. PLoS ONE 2015, 10, e0124375. [Google Scholar]
- Ząbek, T.; Semik, E.; Wnuk, M.; Fornal, A.; Gurgul, A.; Bugno-Poniewierska, M. Epigenetic structure and the role of polymorphism in the shaping of DNA methylation patterns of equine OAS1 locus. J. Appl. Genet. 2015, 56, 231–238. [Google Scholar]
- Braiman, A.; Isakov, N. The role of Crk adaptor proteins in T-cell adhesion and migration. Front. Immunol. 2015, 6, 509. [Google Scholar] [CrossRef]
- Nosrati, M.; Nanaei, H.A.; Javanmard, A.; Esmailizadeh, A. The pattern of runs of homozygosity and genomic inbreeding in world-wide sheep populations. Genomics 2021, 113, 1407–1415. [Google Scholar]
- Addo, S.; Klingel, S.; Thaller, G.; Hinrichs, D. Genetic diversity and the application of runs of homozygosity-based methods for inbreeding estimation in German White-headed Mutton sheep. PLoS ONE 2021, 16, e0250608. [Google Scholar]
- Barani, S.; Nejati-Javaremi, A.; Moradi, M.H.; Moradi-Sharbabak, M.; Gholizadeh, M.; Esfandyari, H. Genome-wide study of linkage disequilibrium, population structure, and inbreeding in Iranian indigenous sheep breeds. PLoS ONE 2023, 18, e0286463. [Google Scholar]
- Peripolli, E.; Munari, D.P.; Silva, M.V.G.B.; Lima, A.L.F.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar]
- Selli, A.; Ventura, R.V.; Fonseca, P.A.; Buzanskas, M.E.; Andrietta, L.T.; Balieiro, J.C.; Brito, L.F. Detection and visualization of heterozygosity-rich regions and runs of homozygosity in worldwide sheep populations. Animals 2021, 11, 2696. [Google Scholar]
- Sanglard, L.P.; Huang, Y.; Gray, K.A.; Linhares, D.C.; Dekkers, J.; Niederwerder, M.C.; Serão, N.V. Further host-genomic characterization of total antibody response to PRRSV vaccination and its relationship with reproductive performance in commercial sows: Genome-wide haplotype and zygosity analyses. Genet. Sel. Evol. 2021, 53, 1–17. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breeds | nROH | Range ROH | NMROH | MGLROH (Mb) | MGPROH (%) | ALROH (Mb) | FROH > 1 Mb | FROH > 8 Mb | FHOM | r (FROH > 1 Mb—FHOM) | r (FROH > 8 Mb—FHOM) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Manipuri | 863 | 13–127 | 1168.62 | 242.88 | 9 | 5.1 | 0.247 | 0.266 | 0.542 | 0.295 | 0.277 |
| Zanskari | 1169 | 17–133 | 1100.94 | 295.5 | 10.94 | 4.89 | 0.285 | 0.189 | 0.546 | 0.261 | 0.357 |
| Bhutia | 961 | 20–116 | 1212.11 | 301.35 | 11.16 | 5.28 | 0.238 | 0.202 | 0.541 | 0.303 | 0.339 |
| Spiti | 1083 | 26–154 | 1139.23 | 409.4 | 15.16 | 5.13 | 0.288 | 0.2 | 0.553 | 0.265 | 0.353 |
| Kathiawari | 630 | 18–147 | 1286.31 | 330.32 | 12.23 | 5.5 | 0.28 | 0.283 | 0.561 | 0.281 | 0.278 |
| Marwari | 574 | 22–111 | 1018.45 | 268.7 | 9.95 | 4.65 | 0.284 | 0.219 | 0.545 | 0.261 | 0.326 |
| Thoroughbred | 164 | 18–114 | 1350.84 | 346.63 | 12.84 | 5.71 | 0.214 | 0.37 | 0.555 | 0.341 | 0.185 |
| Breeds | Manipuri | Zanskari | Bhutia | Spiti | Kathiawari | Marwari | Thoroughbred |
|---|---|---|---|---|---|---|---|
| nROH | 863 | 1169 | 961 | 1083 | 630 | 574 | 164 |
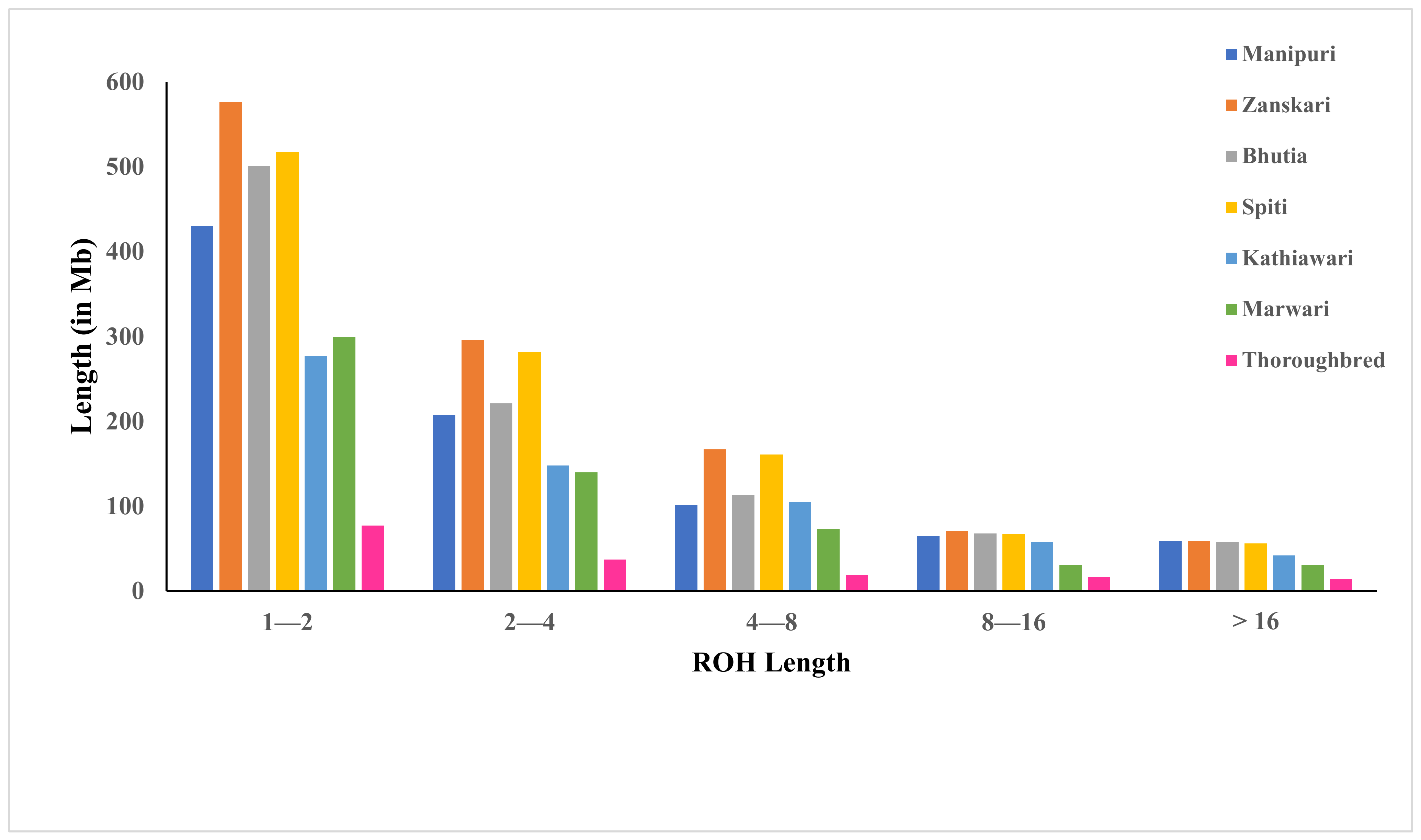
| ROH 1–2 Mb | 430 | 576 | 501 | 517 | 277 | 299 | 77 |
| NROH (percent) | 49.83 | 49.27 | 52.13 | 47.74 | 43.97 | 52.09 | 46.95 |
| Mean length (Mb) ± SD | 1.36 ± 0.28 | 1.37 ± 0.28 | 1.34 ± 0.27 | 1.39 ± 0.28 | 1.39 ± 0.28 | 1.38 ± 0.27 | 1.35 ± 0.27 |
| ROH 2–4 Mb | 208 | 296 | 221 | 282 | 148 | 140 | 37 |
| NROH (percent) | 24.10 | 25.32 | 23.00 | 26.04 | 23.49 | 24.39 | 22.56 |
| Mean length (Mb) ± SD | 2.84 ± 0.60 | 2.79 ± 0.58 | 2.77 ± 0.57 | 2.78 ± 0.59 | 2.85 ± 0.61 | 2.77 ± 0.56 | 2.70 ± 0.52 |
| ROH 4–8 Mb | 101 | 167 | 113 | 161 | 105 | 73 | 19 |
| NROH (percent) | 11.70 | 14.29 | 11.76 | 14.87 | 16.67 | 12.72 | 11.59 |
| Mean length (Mb) ± SD | 5.52 ± 1.08 | 5.47 ± 1.10 | 5.71 ± 1.17 | 5.50 ± 1.03 | 5.58 ± 1.16 | 5.63 ± 1.03 | 5.38 ± 1.05 |
| ROH 8–16 Mb | 65 | 71 | 68 | 67 | 58 | 31 | 17 |
| NROH (percent) | 7.53 | 6.07 | 7.08 | 6.19 | 9.21 | 5.40 | 10.37 |
| Mean length (Mb) ± SD | 10.62 ± 1.97 | 11.20 ± 2.01 | 11.12 ± 2.34 | 10.73 ± 2.23 | 11.10 ± 2.32 | 10.72 ± 1.86 | 11.79 ± 2.27 |
| ROH > 16 Mb | 59 | 59 | 58 | 56 | 42 | 31 | 14 |
| NROH (percent) | 6.84 | 5.05 | 6.04 | 5.17 | 6.67 | 5.40 | 8.54 |
| Mean length (Mb) ± SD | 33.56 ± 22.86 | 40.68 ± 28.18 | 41.25 ± 30.11 | 43.80 ± 28.56 | 33.96 ± 19.20 | 36.32 ± 25.56 | 30.80 ± 17.85 |
| Trait | Marwari | Bhutia | Spiti | Kathiawari | Zanskari | Manipuri | Thoroughbred |
| Health | 13 | 15 | 13 | 16 | 16 | 13 | 17 |
| Growth | 12 | 18 | 15 | 15 | 17 | 14 | 18 |
| Exterior | 2 | 4 | 4 | 1 | 2 | 4 | 0 |
| Reproduction | 7 | 2 | 6 | 3 | 7 | 3 | 5 |
| Performance | 10 | 8 | 9 | 8 | 10 | 8 | 6 |
| Total | 44 | 47 | 47 | 43 | 52 | 42 | 46 |
| Trait | Associated Genes |
| Health | FRZB, XIRP2, MATN1, COL1A2, ARHGAP26, TBC1D22A, CCNG1, GABRA6, TENM2, IGF1, ACAN |
| Growth | OXCT1, HMGA2, MYPN |
| Exterior | MGP, KIT |
| Reproduction | PLCZ1, NXPE3 |
| Performance | SLC16A1, XIRP2, SOX5, CCT5, MRPL44, SERPINE2, CUL3, GIGYF2, INPP5D, MSTN, GRIN2B, SLC16A1, ARL15, DOCK8, HMGA2, DMRT3, MYPN, NXPE3, WDFY2, ATP7B, KCNRG, IGF1, CNTN3, PDZRN3, PPP4R2, GXYLT2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhardwaj, A.; Tandon, G.; Pal, Y.; Sharma, N.K.; Nayan, V.; Soni, S.; Iquebal, M.A.; Jaiswal, S.; Legha, R.A.; Talluri, T.R.; et al. Genome-Wide Single-Nucleotide Polymorphism-Based Genomic Diversity and Runs of Homozygosity for Selection Signatures in Equine Breeds. Genes 2023, 14, 1623. https://doi.org/10.3390/genes14081623
Bhardwaj A, Tandon G, Pal Y, Sharma NK, Nayan V, Soni S, Iquebal MA, Jaiswal S, Legha RA, Talluri TR, et al. Genome-Wide Single-Nucleotide Polymorphism-Based Genomic Diversity and Runs of Homozygosity for Selection Signatures in Equine Breeds. Genes. 2023; 14(8):1623. https://doi.org/10.3390/genes14081623
Chicago/Turabian StyleBhardwaj, Anuradha, Gitanjali Tandon, Yash Pal, Nitesh Kumar Sharma, Varij Nayan, Sonali Soni, Mir Asif Iquebal, Sarika Jaiswal, Ram Avatar Legha, Thirumala Rao Talluri, and et al. 2023. "Genome-Wide Single-Nucleotide Polymorphism-Based Genomic Diversity and Runs of Homozygosity for Selection Signatures in Equine Breeds" Genes 14, no. 8: 1623. https://doi.org/10.3390/genes14081623
APA StyleBhardwaj, A., Tandon, G., Pal, Y., Sharma, N. K., Nayan, V., Soni, S., Iquebal, M. A., Jaiswal, S., Legha, R. A., Talluri, T. R., Bhattacharya, T. K., Kumar, D., Rai, A., & Tripathi, B. N. (2023). Genome-Wide Single-Nucleotide Polymorphism-Based Genomic Diversity and Runs of Homozygosity for Selection Signatures in Equine Breeds. Genes, 14(8), 1623. https://doi.org/10.3390/genes14081623