
Co-Occurrence of Germline Genomic Variants and Copy Number Variations in Hereditary Breast and Colorectal Cancer Patients

 , , ,
, , ,
Abstract
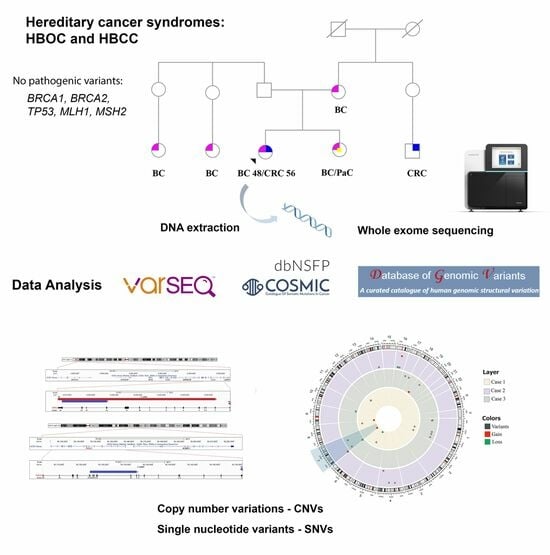
1. Background
2. Methods
2.1. Patients
2.2. DNA Extraction, Whole Exome Sequencing (WES), and Data Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Dash, C.; Lu, J.; Parikh, V.; Wathen, S.; Shah, S.; Shah Chaudhari, R.; Adams-Campbell, L. Disparities in colorectal cancer screening among breast and prostate cancer survivors. Cancer Med. 2021, 10, 1448–1456. [Google Scholar] [CrossRef]
- Halamkova, J.; Kazda, T.; Pehalova, L.; Gonec, R.; Kozakova, S.; Bohovicova, L.; Krakorova, D.A.; Slaby, O.; Demlova, R.; Svoboda, M.; et al. Second primary malignancies in colorectal cancer patients. Sci. Rep. 2021, 11, 2759. [Google Scholar] [CrossRef]
- Akcay, I.M.; Celik, E.; Agaoglu, N.B.; Alkurt, G.; Kizilboga Akgun, T.; Yildiz, J.; Enc, F.; Kir, G.; Canbek, S.; Kilic, A.; et al. Germline pathogenic variant spectrum in 25 cancer susceptibility genes in Turkish breast and colorectal cancer patients and elderly controls. Int. J. Cancer 2021, 148, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R. Hereditary breast and ovarian cancer (HBOC): Review of its molecular characteristics, screening, treatment, and prognosis. Breast Cancer 2021, 28, 1167–1180. [Google Scholar] [CrossRef]
- Sheehan, M.; Heald, B.; Yanda, C.; Kelly, E.D.; Grobmyer, S.; Eng, C.; Kalady, M.; Pederson, H. Investigating the Link between Lynch Syndrome and Breast Cancer. Eur. J. Breast Health 2020, 16, 106–109. [Google Scholar] [CrossRef]
- Schwartz, C.J.; da Silva, E.M.; Marra, A.; Gazzo, A.M.; Selenica, P.; Rai, V.K.; Mandelker, D.; Pareja, F.; Misyura, M.; D’Alfonso, T.M.; et al. Morphologic and Genomic Characteristics of Breast Cancers Occurring in Individuals with Lynch Syndrome. Clin. Cancer Res. 2022, 28, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Win, A.K.; Jenkins, M.A.; Dowty, J.G.; Antoniou, A.C.; Lee, A.; Giles, G.G.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Ahnen, D.J.; et al. Prevalence and penetrance of major genes and polygenes for colorectal cancer. Cancer Epidemiol. Biomark. Prev. 2017, 26, 404–412. [Google Scholar] [CrossRef]
- Biller, L.H.; Creedon, S.A.; Klehm, M.; Yurgelun, M.B. Lynch Syndrome-Associated Cancers Beyond Colorectal Cancer. Gastrointest Endosc. Clin. 2022, 32, 75–93. [Google Scholar] [CrossRef] [PubMed]
- McBride, K.A.; Ballinger, M.L.; Killick, E.; Kirk, J.; Tattersall, M.H.N.; Eeles, R.A.; Thomas, D.M.; Mitchell, G. Li-Fraumeni syndrome: Cancer risk assessment and clinical management. Nat. Rev. Clin. Oncol. 2014, 11, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Soeda, S.; Endo, Y.; Okabe, C.; Sato, T.; Kamo, N.; Ueda, M.; Kojima, M.; Furukawa, S.; Nishigori, H.; et al. Rare Hereditary Gynecological Cancer Syndromes. Int. J. Mol. Sci. 2022, 23, 1563. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.C.; Lisboa, B.C.; Figueiredo, M.C.; Torrezan, G.T.; Santos, E.M.; Krepischi, A.C.; Rossi, B.M.; Achatz, M.I.; Carraro, D.M. Hereditary breast and ovarian cancer: Assessment of point mutations and copy number variations in Brazilian patients. BMC Med. Genet. 2014, 15, 55. [Google Scholar] [CrossRef]
- Villacis, R.A.R.; Abreu, F.B.; Miranda, P.M.; Domingues, M.A.C.; Carraro, D.M.; Santos, E.M.M.; Andrade, V.P.; Rossi, B.M.; Achatz, M.I.; Rogatto, S.R. ROBO1 deletion as a novel germline alteration in breast and colorectal cancer patients. Tumour Biol. 2016, 37, 3145–3153. [Google Scholar] [CrossRef] [PubMed]
- Villacis, R.A.R.; Basso, T.R.; Canto, L.M.; Pinheiro, M.; Santiago, K.M.; Giacomazzi, J.; de Paula, C.A.A.; Carraro, D.M.; Ashton-Prolla, P.; Achatz, M.I.; et al. Rare germline alterations in cancer-related genes associated with the risk of multiple primary tumor development. J. Mol. Med. 2017, 95, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Boujemaa, M.; Hamdi, Y.; Mejri, N.; Romdhane, L.; Ghedira, K.; Bouaziz, H.; El Benna, H.; Labidi, S.; Dallali, H.; Jaidane, O.; et al. Germline copy number variations in BRCA1/2 negative families: Role in the molecular etiology of hereditary breast cancer in Tunisia. PLoS ONE 2021, 16, e0245362. [Google Scholar] [CrossRef] [PubMed]
- Krepischi, A.C.; Achatz, M.I.; Santos, E.M.; Costa, S.S.; Lisboa, B.C.; Brentani, H.; Santos, T.M.; Gonçalves, A.; Nóbrega, A.F.; Pearson, P.L.; et al. Germline DNA copy number variation in familial and early-onset breast cancer. Breast Cancer Res. 2012, 14, R24. [Google Scholar] [CrossRef]
- Pylkäs, K.; Vuorela, M.; Otsukka, M.; Kallioniemi, A.; Jukkola-Vuorinen, A.; Winqvist, R. Rare copy number variants observed in hereditary breast cancer cases disrupt genes in estrogen signaling and TP53 tumor suppression network. PLoS Genet. 2012, 8, e1002734. [Google Scholar] [CrossRef]
- Murphy, C.C.; Gerber, D.E.; Pruitt, S.L. Prevalence of Prior Cancer Among Persons Newly Diagnosed with Cancer: An Initial Report from the Surveillance, Epidemiology, and End Results Program. JAMA Oncol. 2018, 4, 832–836. [Google Scholar] [CrossRef]
- Vogt, A.; Schmid, S.; Heinimann, K.; Frick, H.; Herrmann, C.; Cerny, T.; Omlin, A. Multiple primary tumours: Challenges and approaches, a review. ESMO Open 2017, 2, e000172. [Google Scholar] [CrossRef]
- La Francis, I.E.; Cooper, R.B. Second primary malignancies associated with primary female breast cancer: A review of the Danbury Hospital experience. Conn. Med. 1992, 56, 411–414. [Google Scholar]
- Soerjomataram, I.; Louwman, W.J.; de Vries, E.; Lemmens, V.E.; Klokman, W.J.; Coebergh, J.W. Primary malignancy afer primary female breast cancer in the south of the Netherlands, 1972–2001. Breast Cancer Res. Treat. 2005, 93, 91–95. [Google Scholar] [CrossRef]
- Naseem, H.; Boylan, J.; Speake, D.; Leask, K.; Shenton, A.; Lalloo, F.; Hill, J.; Trump, D.; Evans, D.G. Inherited association of breast and colorectal cancer: Limited role of CHEK2 compared with high-penetrance genes. Clin. Genet. 2006, 70, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Phelan, C.M.; Iqbal, J.; Lynch, H.T.; Lubinski, J.; Gronwald, J.; Moller, P.; Ghadirian, P.; Foulkes, W.D.; Armel, S.; Eisen, A.; et al. Incidence of colorectal cancer in brca1 and BRCA2 mutation carriers: Results from a follow-up study. Br. J. Cancer 2014, 110, 530–534. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Naslavsky, M.S.; Yamamoto, G.L.; de Almeida, T.F.; Ezquina, S.A.M.; Sunaga, D.Y.; Pho, N.; Bozoklian, D.; Sandberg, T.O.M.; Brito, L.A.; Lazar, M.; et al. Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum. Mutat. 2017, 38, 751–763. [Google Scholar] [CrossRef]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed]
- Meijers-Heijboer, H.; Wijnen, J.; Vasen, H.; Wasielewski, M.; Wagner, A.; Hollestelle, A.; Elstrodt, F.; van den Bos, R.; de Snoo, A.; Fat, G.T.; et al. The CHEK2 1100delC mutation identifies families with a hereditary breast and colorectal cancer phenotype. Am. J. Hum. Genet. 2003, 72, 1308–1314. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, J.; Kassem, M.; Agnese, D.; Pilarski, R.; Ramaswamy, B.; Sweet, K.; Sardesai, S. Concurrent germline BRCA1, BRCA2, and CHEK2 pathogenic variants in hereditary breast cancer: A case series. Breast Cancer Res. Treat. 2021, 2, 569–575. [Google Scholar] [CrossRef]
- Beltrami, C.M.; Canto, L.M.; Villacis, R.A.R.; Petersen, A.H.; Aagaard, M.M.; Cury, S.S.; Formiga, M.N.C.; Junior, S.A.; Rogatto, R.R. The repertoire of germline variants in patients with early-onset rectal cancer. Cancer Commun. 2022, 5, 481–485. [Google Scholar] [CrossRef]
- Megid, T.B.C.; Barros-Filho, M.C.; Pisani, J.P.; Achatz, M.I. Double heterozygous pathogenic variants prevalence in a cohort of patients with hereditary breast cancer. Front. Oncol. 2022, 12, 873395. [Google Scholar] [CrossRef]
- Agaoglu, N.B.; Doganay, L. Concurrent pathogenic variations in patients with hereditary cancer syndromes. Eur. J. Med. Genet. 2021, 64, 104366. [Google Scholar] [CrossRef]
- Lin, D.I.; Huang, R.S.P.; Mata, D.A.; Decker, B.; Danziger, N.; Lechpammer, M.; Hiemenz, M.; Ramkissoon, S.H.; Ross, J.S.; Elvin, J.A. Clinicopathological and genomic characterization of BCORL1-driven high-grade endometrial stromal sarcomas. Mod. Pathol. 2021, 34, 2200–2210. [Google Scholar] [CrossRef]
- Min, J.Y.; Lee, G.H.; Khanal, T.; Jin, S.W.; Lee, S.Y.; Kim, H.G.; Hyon, J.Y.; Chung, Y.H.; Ha, S.K.; Han, E.H.; et al. Upregulation of CYP1B1 by hypoxia is mediated by ERα activation in breast cancer cells. Am. J. Cancer Res. 2022, 12, 2798–2816. [Google Scholar]
- He, X.F.; Wei, J.; Liu, Z.Z.; Xie, J.J.; Wang, W.; Du, Y.P.; Chen, Y.; Si, H.Q.; Liu, Q.; Wu, L.X.; et al. Association between CYP1A2 and CYP1B1 polymorphisms and colorectal cancer risk: A meta-analysis. PLoS ONE 2014, 9, e100487. [Google Scholar]
- Vishnubalaji, R.; Alajez, N.M. Epigenetic regulation of triple negative breast cancer (TNBC) by TGF-β signaling. Sci. Rep. 2021, 11, 15410. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, F.; Wu, J.; Zhang, T.; Liu, F.; Mao, Y.; Hua, D. Expression of CYP1B1 and B7-H3 significantly correlates with poor prognosis in colorectal cancer patients. Int. J. Clin. Exp. Pathol. 2018, 11, 2654–2664. [Google Scholar] [PubMed]
- Solomon, J.P.; Benayed, R.; Hechtman, J.F.; Ladanyi, M. Identifying patients with NTRK fusion cancer. Ann. Oncol. 2019, 30, viii16–viii22. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open. 2016, 1, e000023. [Google Scholar] [CrossRef]
- Lieber, S.; Reinartz, S.; Raifer, H.; Finkernagel, F.; Dreyer, T.; Bronger, H.; Jansen, J.M.; Wagner, U.; Worzfeld, T.; Müller, R.; et al. Prognosis of ovarian cancer is associated with effector memory CD8+ T cell accumulation in ascites, CXCL9 levels and activation-triggered signal transduction in T cells. Oncoimmunology 2018, 7, e1424672. [Google Scholar] [CrossRef]
- Perret, R.; Escuriol, J.; Velasco, V.; Mayeur, L.; Soubeyran, I.; Delfour, C.; Aubert, S.; Polivka, M.; Karanian, M.; Meurgey, A.; et al. NFATc2-rearranged sarcomas: Clinicopathologic, molecular, and cytogenetic study of 7 cases with evidence of AGGRECAN as a novel diagnostic marker. Mod. Pathol. 2020, 33, 1930–1944. [Google Scholar] [CrossRef]
- Lang, T.; Ding, X.; Kong, L.; Zhou, X.; Zhang, Z.; Ju, H.; Ding, S. NFATC2 is a novel therapeutic target for colorectal cancer stem cells. Onco Targets Ther. 2018, 11, 6911–6924. [Google Scholar] [CrossRef]
- Vaughn, C.P.; Hart, K.J.; Samowitz, W.S.; Swensen, J.J. Avoidance of pseudogene interference in the detection of 3’ deletions in PMS2. Hum. Mutat. 2011, 32, 1063–1071. [Google Scholar] [CrossRef]
- Blount, J.; Prakash, A. The Changing Landscape of Lynch Syndrome due to PMS2 Mutations. Clin. Genet. 2018, 94, 61–69. [Google Scholar] [CrossRef]
- Herman, D.S.; Smith, C.; Liu, C.; Vaughn, C.P.; Palaniappan, S.; Pritchard, C.C.; Shirts, B.H. Efficient detection of copy number mutations in PMS2 exons with a close homolog. J. Mol. Diagn. 2018, 20, 512–521. [Google Scholar] [CrossRef]
- Niessen, R.C.; Kleibeuker, J.H.; Jager, P.O.J.; Sijmons, R.H.; Hofstra, R.M.W. Getting rid of the PMS2 pseudogenes: Mission impossible? Hum. Mutat. 2007, 28, 414. [Google Scholar] [CrossRef] [PubMed]
- Pös, O.; Radvanszky, J.; Buglyó, G.; Pös, Z.; Rusnakova, D.; Nagy, B.; Szemes, T. DNA copy number variation: Main characteristics, evolutionary significance, and pathological aspects. Biomed. J. 2021, 44, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Olsen, M.F.; Lavik, L.A.S.; Vold, T.; Drabløs, F.; Sjursen, W. Detecting copy number variation in next generation sequencing data from diagnostic gene panels. BMC Med. Genomics 2021, 14, 214. [Google Scholar] [CrossRef] [PubMed]
- Fragoso-Ontiveros, V.; De la Fuente-Hernandez, M.A.; Gonzalez-Osnaya, V.; Gamez-Rosales, M.; Perez-Montiel, M.D.; Isla-Ortiz, D.; Cantu-De Leon, D.F.; Alvarez-Gomez, R.M. A Pathogenic Variant Reclassified to the Pseudogene PMS2P1 in a Patient with Suspected Hereditary Cancer. Int. J. Mol. Sci. 2023, 24, 1398. [Google Scholar] [CrossRef]
- Roberts, M.E.; Jackson, S.A.; Susswein, L.R.; Zeinomar, N.; Ma, X.; Marshall, M.L.; Stettner, A.R.; Milewski, B.; Xu, Z.; Solomon, B.D.; et al. MSH6 and PMS2 germline pathogenic variants implicated in Lynch syndrome are associated with breast cancer. Genet. Med. 2018, 20, 1167–1174. [Google Scholar] [CrossRef]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Almeida, E.G.; Sanguero, I.; et al. Germline mutations in the proof-reading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 2, 136–144. [Google Scholar] [CrossRef]
- Chubb, D.; Broderick, P.; Dobbins, S.E.; Frampton, M.; Kinnersley, B.; Penegar, S.; Price, A.; Ma, Y.P.; Sherborne, A.L.; Palles, C.; et al. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat. Commun. 2016, 7, 11883. [Google Scholar] [CrossRef]
- Spier, I.; Holzapfel, S.; Altmüller, J.; Zhao, B.; Horpaopan, S.; Vogt, S.; Chen, S.; Morak, M.; Raeder, S.; Kayser, K.; et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int. J. Cancer 2015, 137, 320–331. [Google Scholar] [CrossRef]
- Perea, J.; García, J.L.; Pérez, J.; Rueda, D.; Arriba, M.; Rodríguez, Y.; Urioste, M.; González-Sarmiento, R. NOMO-1 gene is deleted in early-onset colorectal cancer. Oncotarget 2017, 8, 24429–24436. [Google Scholar] [CrossRef]
- Pérez-García, J.; Martel-Martel, A.; García-Vallés, P.; Corchete, L.A.; García, J.L.; Gestoso-Uzal, N.; Vidal-Tocino, R.; Blanco, Ó.; Méndez, L.; Sánchez-Martín, M.; et al. Recurrent NOMO1 Gene Deletion Is a Potential Clinical Marker in Early-Onset Colorectal Cancer and Is Involved in the Regulation of Cell Migration. Cancers 2022, 14, 4029. [Google Scholar] [CrossRef] [PubMed]
- Meng, T.C.; Fukada, T.; Tonks, N.K. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell 2002, 9, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Cao, Y.; Wang, Y.; Li, W.; Liu, X.; Lv, Y.; Li, X.; Mi, J. Cysteine transporter SLC3A1 promotes breast cancer tumorigenesis. Theranostics 2017, 7, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, L.Y.; Arsene, D.; Mato, J.M.; Lu, S.C. Methionine adenosyltransferases in cancers: Mechanisms of dysregulation and implications for therapy. Exp. Biol. Med. 2018, 243, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Biswas, K.; Sommers, J.A.; Thompson, H.; Awate, S.; Nicolae, C.M.; Thakar, T.; Moldovan, G.L.; Shoemaker, R.H.; Sharan, S.K.; et al. WRN helicase safeguards deprotected replication forks in BRCA2-mutated cancer cells. Nat. Commun. 2021, 12, 6561. [Google Scholar] [CrossRef]
- Frau, M.; Feo, F.; Pascale, R.M. Pleiotropic effects of methionine adenosyltransferases deregulation as determinants of liver cancer progression and prognosis. J. Hepatol. 2013, 59, 830–841. [Google Scholar] [CrossRef]
- Frigola, J.; Muñoz, M.; Clark, S.J.; Moreno, V.; Capellà, G.; Peinado, M.A. Hypermethylation of the prostacyclin synthase (PTGIS) promoter is a frequent event in colorectal cancer and associated with aneuploidy. Oncogene 2005, 24, 7320–7326. [Google Scholar] [CrossRef]
- Dai, D.; Chen, B.; Feng, Y.; Wang, W.; Jiang, Y.; Huang, H.; Liu, J. Prognostic value of prostaglandin I2 synthase and its correlation with tumor-infiltrating immune cells in lung cancer, ovarian cancer, and gastric cancer. Aging 2020, 12, 9658–9685. [Google Scholar] [CrossRef]
- Lichao, S.; Liang, P.; Chunguang, G.; Fang, L.; Zhihua, Y.; Yuliang, R. Overexpression of PTGIS could predict liver metastasis and is correlated with poor prognosis in colon cancer patients. Pathol. Oncol. Res. 2012, 18, 563–569. [Google Scholar] [CrossRef]
- Iglesias-Pedraz, J.M.; Fossatti-Jara, D.M.; Valle-Riestra-Felice, V.; Cruz-Visalaya, S.R.; Ayala Felix, J.A.; Comai, L. WRN modulates translation by influencing nuclear mRNA export in HeLa cancer cells. BMC Mol. Cell Biol. 2020, 21, 71. [Google Scholar] [CrossRef]
- Yang, L.; Wang, G.; Zhao, X.; Ye, S.; Shen, P.; Wang, W.; Zheng, S. A Novel WRN Frameshift Mutation Identified by Multiplex Genetic Testing in a Family with Multiple Cases of Cancer. PLoS ONE 2015, 10, e0133020. [Google Scholar] [CrossRef]
- Zimmer, K.; Puccini, A.; Xiu, J.; Baca, Y.; Spizzo, G.; Lenz, H.J.; Battaglin, F.; Goldberg, R.M.; Grothey, A.; Shields, A.F.; et al. WRN-Mutated Colorectal Cancer Is Characterized by a Distinct Genetic Phenotype. Cancers 2020, 12, 1319. [Google Scholar] [CrossRef] [PubMed]
- Norouzi, M.; Shafiei, M.; Abdollahi, Z.; Miar, P.; Galehdari, H.; Emami, M.H.; Zeinalian, M.; Tabatabaiefar, M.A. WRN Germline Mutation Is the Likely Inherited Etiology of Various Cancer Types in One Iranian Family. Front. Oncol. 2021, 11, 648649. [Google Scholar] [CrossRef] [PubMed]
- Lietman, C.D.; McKean, M. Targeting GNAQ/11 through PKC inhibition in uveal melanoma. Cancer Gene Ther. 2022, 29, 1809–1813. [Google Scholar] [CrossRef]
- Lee, S.H.; Jung, S.H.; Kim, T.M.; Rhee, J.K.; Park, H.C.; Kim, M.S.; Kim, S.S.; An, C.H.; Lee, S.H.; Chung, Y.J. Whole-exome sequencing identified mutational profiles of high-grade colon adenomas. Oncotarget 2017, 8, 6579–6588. [Google Scholar] [CrossRef] [PubMed]
- Waszak, S.M.; Northcott, P.A.; Buchhalter, I.; Robinson, G.W.; Sutter, C.; Groebner, S.; Grund, K.B.; Brugières, L.; Jones, D.T.W.; Pajtler, K.W.; et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: A retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 2018, 19, 785–798. [Google Scholar] [CrossRef]
- Yoshida, M.; Nakabayashi, K.; Yang, W.; Sato-Otsubo, A.; Tsujimoto, S.I.; Ogata-Kawata, H.; Kawai, T.; Ishiwata, K.; Sakamoto, M.; Okamura, K.; et al. Prevalence of pathogenic variants in cancer-predisposing genes in second cancer after childhood solid cancers. Cancer Med. 2023, 12, 11264–11273. [Google Scholar] [CrossRef]
- Poliani, L.; Greco, L.; Barile, M.; Dal Buono, A.; Bianchi, P.; Basso, G.; Giatti, V.; Genuardi, M.; Malesci, A.; Laghi, L.; et al. Canonical and uncanonical pathogenic germline variants in colorectal cancer patients by next-generation sequencing in a European referral center. ESMO Open 2022, 7, 100607. [Google Scholar] [CrossRef]
- Chang, N.C.; Sincennes, M.C.; Chevalier, F.P.; Brun, C.E.; Lacaria, M.; Segalés, J.; Muñoz-Cánoves, P.; Ming, H.; Rudnicki, M.A. The Dystrophin Glycoprotein Complex Regulates the Epigenetic Activation of Muscle Stem Cell Commitment. Cell Stem Cell 2018, 22, 755–768. [Google Scholar] [CrossRef]
- Zeng, K.; Wu, Y.; Wang, C.; Wang, S.; Sun, H.; Zou, R.; Sun, G.; Song, H.; Liu, W.; Sun, N.; et al. ASH2L is involved in promotion of endometrial cancer progression via upregulation of PAX2 transcription. Cancer Sci. 2020, 111, 2062–2077. [Google Scholar] [CrossRef]
- Qi, J.; Huo, L.; Zhu, Y.T.; Zhu, Y.J. Absent, small or homeotic 2-like protein (ASH2L) enhances the transcription of the estrogen receptor α gene through GATA-binding protein 3 (GATA3). J. Biol. Chem. 2014, 289, 31373–31381. [Google Scholar] [CrossRef] [PubMed]
- Mungamuri, S.K.; Wang, S.; Manfredi, J.J.; Gu, W.; Aaronson, S.A. Ash2L enables P53-dependent apoptosis by favoring stable transcription pre-initiation complex formation on its pro-apoptotic target promoters. Oncogene 2015, 34, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Huang, W.; Yang, Y.; Qiu, R.; Zeng, Y.; Hou, Y.; Sun, G.; Shi, H.; Leng, S.; Feng, D.; et al. GATA3 recruits UTX for gene transcriptional activation to suppress metastasis of breast cancer. Cell Death Dis. 2019, 10, 832. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Patient | Clinical Features | Relatives with Cancer |
|---|---|---|
| 1 | BC (64—left, T1N0M0, IDC-NE) | Sister (BC, 68) |
| BC (64—right, TxN1M0, adenocarcinoma, NI) | ||
| CRC (71, TxNxMx, rectal carcinoma, NI) | ||
| 2 | BC (55—left, T2N2aM0, IDC-NE—ER+/PR−/HER2−, surgery + CT + RT) | Sister (BC, 40 and CRC, 48) |
| BC (55—right, T2N0M0, IDC-NE—ER+/PR+/HER2+, surgery + CT + RT) | Sister (thyroid cancer, 45) | |
| CRC (57, T3N2M0, TA, surgery + CT) | Maternal aunt (bone cancer, 52) | |
| 3 | BC (48—right, TxN0M0, IDC-NE, surgery + CT + RT) CRC (56, T3N2M0, TA, surgery + CT) | Mother (bilateral BC, NI) |
| Sister (BC, NI and pancreas cancer, NI) | ||
| Maternal cousin (CRC, NI) | ||
| Paternal cousin (BC, NI) | ||
| Paternal cousin (BC, NI) |
| SINGLE NUCLEOTIDE VARIANTS | |||||||||
| Patient | Gene | cDNA | Protein | Type/Effect | dbNSFP * | GnomAD Variant Frequency | NCBI RefSeq | dbSNP 154 v2, NCBI | ACMG/ClinVar |
| 1 | CYP1B1 | c.1159G > A | p.Glu387Lys | Missense | 4 | 0.00029 | NM_000104.4 | rs55989760 | LP/LP |
| NTRK1 | c.16C > T | p.Arg6Trp | Missense | 4 | 0.00325 | NM_001012331.2 | rs201472270 | VUS/- | |
| NFATC2 | c.1952G > A | p.Arg651His | Missense | 4 | 0.00006 | NM_173091.4 | rs148347040 | VUS/- | |
| (+47 VUS) | |||||||||
| 2 | SLC3A1 | c.1400T > C | p.Met467Thr | Missense | 5 | 0.00250 | NM_000341.4 | rs121912691 | VUS/LP |
| MAT1A | c.826dupG | p.Ala276Glyfs*76 | LoF | - | 0.00001 | NM_000429.3 | rs763178849 | LP/- | |
| PTGIS | c.824G > A | p.Arg275Gln | Missense | 3 | 0.00098 | NM_000961.4 | rs61734270 | VUS/LP | |
| WRN | c.95A > G | p.Lys32Arg | Missense | 3 | 0.00305 | NM_000553.6 | rs34477820 | VUS/- | |
| (+58 VUS) | |||||||||
| 3 | GNAQ | c.286A > T | p.Thr96Ser | Missense | 3 | 0.00440 | NM_002072.5 | rs753716491 | VUS/- |
| PTCH1 | c.140G > T | p.Arg47Leu | Missense | 2 | 0 | NM_000264.5 | rs775408408 | VUS/- | |
| (+31 VUS) | |||||||||
| COPY NUMBER VARIATIONS | |||||||||
| Patient | Gene | Chrom | Start | End | Type | Other Genes | ACMG/ClinVar | ||
| 1 | PMS2 | 7 | 5764955 | 6045662 | Loss | CCZ1, OCM, RNF216, RSPH10B | VUS/VUS | ||
| POLE2 | 14 | 50154854 | 50160008 | Gain | KLHDC1 | VUS/- | |||
| NOMO1 | 16 | 14946287 | 14959012 | Loss | - | VUS/- | |||
| (+209 CNVs) | |||||||||
| 2 | PMS2 | 7 | 6013030 | 6022622 | Gain | - | VUS/- | ||
| (+16 CNVs) | |||||||||
| 3 | PMS2 | 7 | 6048628 | 6049129 | Loss | AIMP2 | VUS/P | ||
| ASH2L | 8 | 37914599 | 37963256 | Loss | EIF4EBP1 | VUS/- | |||
| POLE2 | 14 | 50120873 | 50131392 | Gain | - | VUS/- | |||
| (+147 CNVs) | |||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Côrtes, L.; Basso, T.R.; Villacis, R.A.R.; Souza, J.d.S.; Jørgensen, M.M.A.; Achatz, M.I.; Rogatto, S.R. Co-Occurrence of Germline Genomic Variants and Copy Number Variations in Hereditary Breast and Colorectal Cancer Patients. Genes 2023, 14, 1580. https://doi.org/10.3390/genes14081580
Côrtes L, Basso TR, Villacis RAR, Souza JdS, Jørgensen MMA, Achatz MI, Rogatto SR. Co-Occurrence of Germline Genomic Variants and Copy Number Variations in Hereditary Breast and Colorectal Cancer Patients. Genes. 2023; 14(8):1580. https://doi.org/10.3390/genes14081580
Chicago/Turabian StyleCôrtes, Luiza, Tatiane Ramos Basso, Rolando André Rios Villacis, Jeferson dos Santos Souza, Mads Malik Aagaard Jørgensen, Maria Isabel Achatz, and Silvia Regina Rogatto. 2023. "Co-Occurrence of Germline Genomic Variants and Copy Number Variations in Hereditary Breast and Colorectal Cancer Patients" Genes 14, no. 8: 1580. https://doi.org/10.3390/genes14081580
APA StyleCôrtes, L., Basso, T. R., Villacis, R. A. R., Souza, J. d. S., Jørgensen, M. M. A., Achatz, M. I., & Rogatto, S. R. (2023). Co-Occurrence of Germline Genomic Variants and Copy Number Variations in Hereditary Breast and Colorectal Cancer Patients. Genes, 14(8), 1580. https://doi.org/10.3390/genes14081580