
Possible Effects of Uremic Toxins p-Cresol, Indoxyl Sulfate, p-Cresyl Sulfate on the Development and Progression of Colon Cancer in Patients with Chronic Renal Failure

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
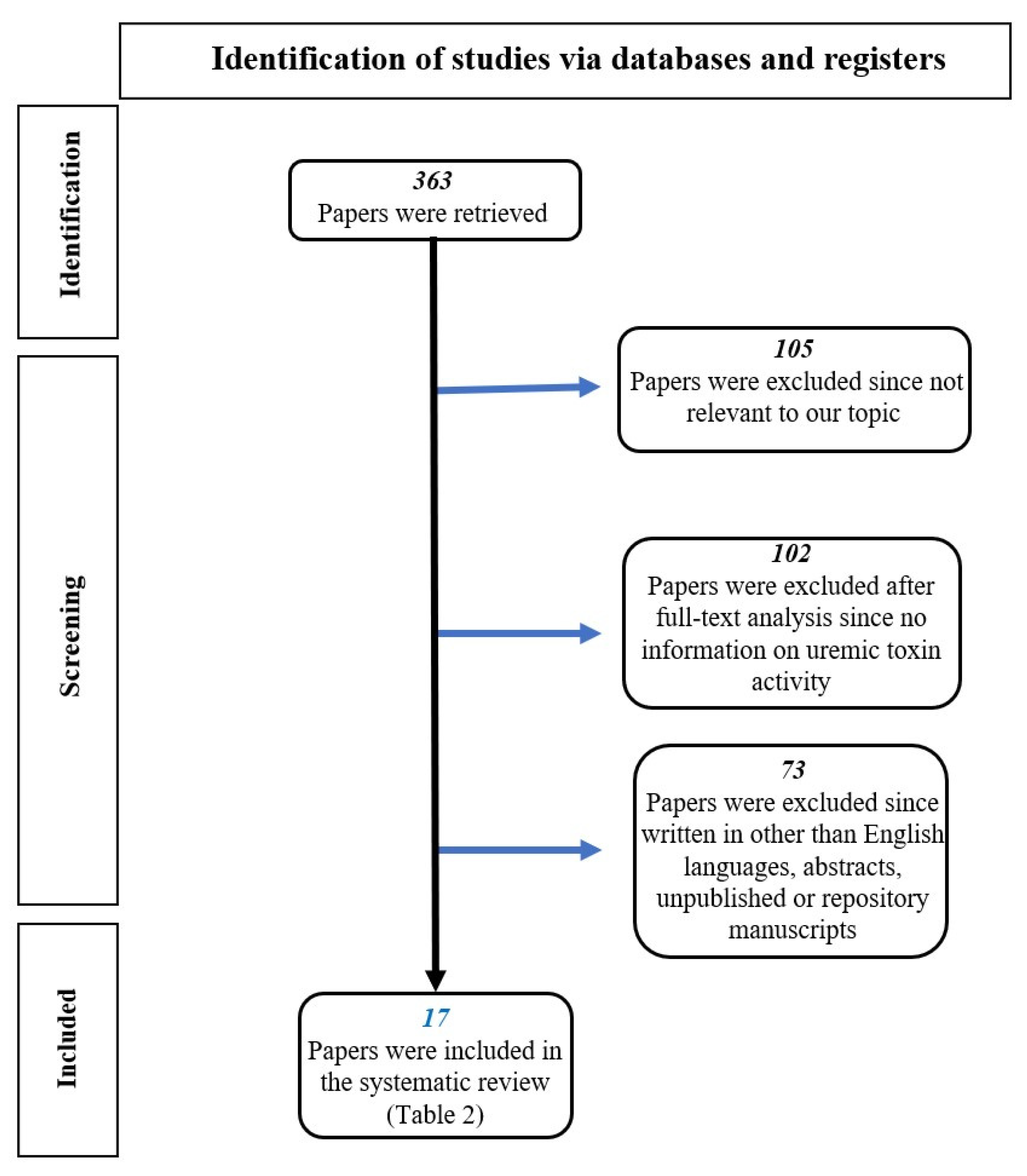
2.1. Conduct of Review
2.2. Search Strategy and Study Selection
3. Results
3.1. Intestinal Dysbiosis in CKD
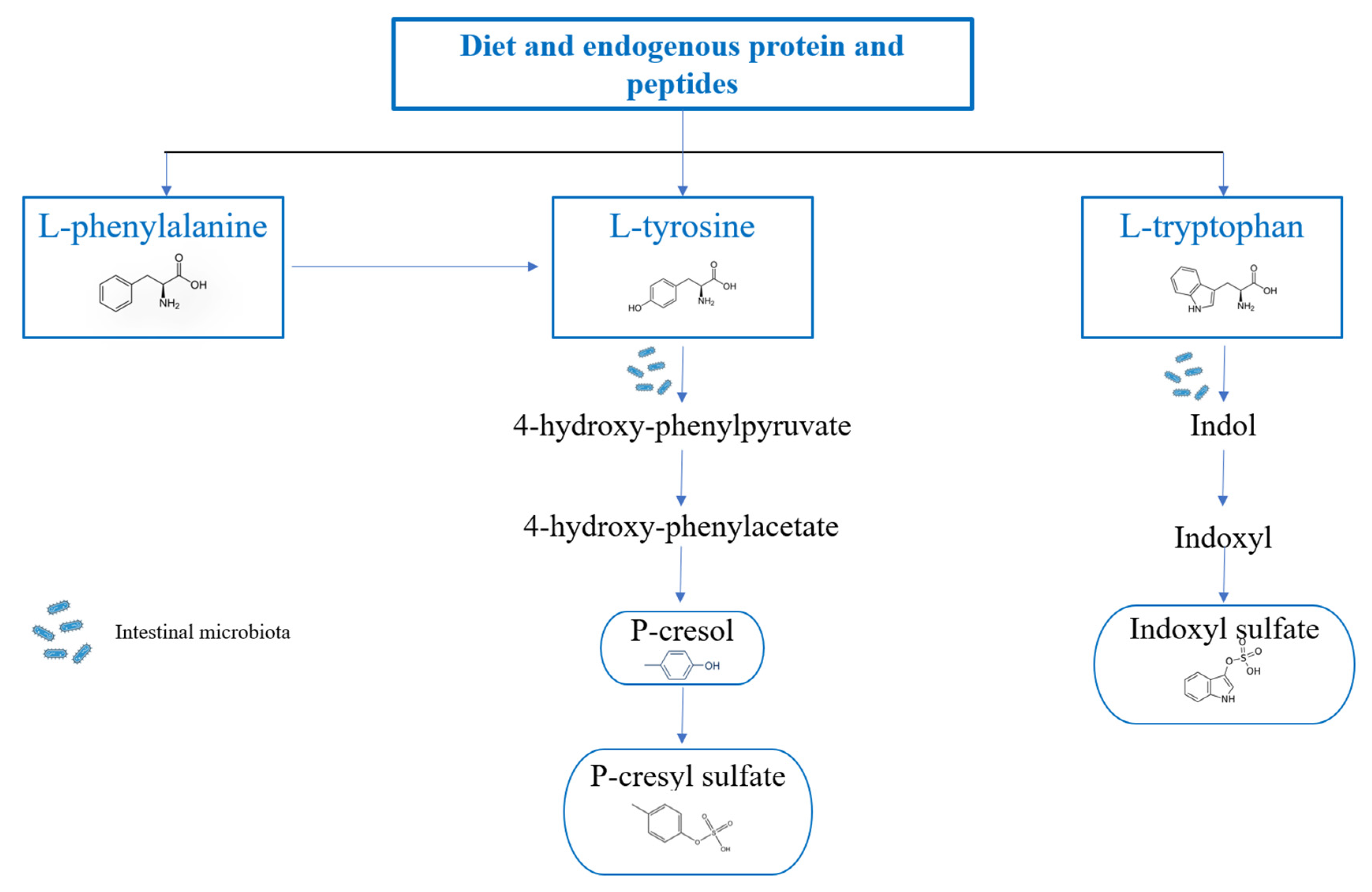
3.2. Metabolism of p-Cresol, p-Cresyl Sulfate and Indoxyl Sulfate
3.3. Uremic Toxins and Cancers
3.4. Association of CKD Dysbiosis and Colorectal Cancer: In It a Concrete Hypothesis?
3.5. Chronic Intestinal Inflammation Induced by IS
3.6. Immune Dysfunction Induced by p-CS and IS
3.7. Alteration of the Permeability of the Intestinal Epithelium Induced by IS
3.8. Increase in ROS Production Induced by IS
3.9. DNA Damage in Enterocytes and p-C
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2017, 11, 1–10. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-Bacterial Mutualism in the Human Intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef]
- Hobby, G.P.; Karaduta, O.; Dusio, G.F.; Singh, M.; Zybailov, B.L.; Arthur, J.M. Chronic kidney disease and the gut microbiome. Am. J. Physiol. Physiol. 2019, 316, F1211–F1217. [Google Scholar] [CrossRef]
- Ramezani, A.; Raj, D.S. The Gut Microbiome, Kidney Disease, and Targeted Interventions. J. Am. Soc. Nephrol. 2014, 25, 657–670. [Google Scholar] [CrossRef]
- Kang, J.Y. The gastrointestinal tract in uremia. Dig. Dis. Sci. 1993, 38, 257–268. [Google Scholar] [CrossRef]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The Uremic Toxicity of Indoxyl Sulfate and p-Cresyl Sulfate: A Systematic Review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef]
- Saumoy, M.; Jesudian, A.B.; Aden, B.; Serur, D.; Sundararajan, S.; Sivananthan, G.; Gambarin-Gelwan, M. High prevalence of colon adenomas in end-stage kidney disease patients on hemodialysis undergoing renal transplant evaluation. Clin. Transplant. 2016, 30, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Shafi, T.; Meyer, T.W.; Hostetter, T.H.; Melamed, M.L.; Parekh, R.S.; Hwang, S.; Banerjee, T.; Coresh, J.; Powe, N.R. Free Levels of Selected Organic Solutes and Cardiovascular Morbidity and Mortality in Hemodialysis Patients: Results from the Retained Organic Solutes and Clinical Outcomes (ROSCO) Investigators. PLoS ONE 2015, 10, e0126048. [Google Scholar] [CrossRef] [PubMed]
- Rapa, S.F.; Prisco, F.; Popolo, A.; Iovane, V.; Autore, G.; Di Iorio, B.R.; Dal Piaz, F.; Paciello, O.; Nishijima, F.; Marzocco, S. Pro-Inflammatory Effects of Indoxyl Sulfate in Mice: Impairment of Intestinal Homeostasis and Immune Response. Int. J. Mol. Sci. 2021, 22, 1135. [Google Scholar] [CrossRef]
- Huang, Y.; Zhou, J.; Wang, S.; Xiong, J.; Chen, Y.; Liu, Y.; Xiao, T.; Li, Y.; He, T.; Li, Y.; et al. Indoxyl sulfate induces intestinal barrier injury through IRF1-DRP1 axis-mediated mitophagy impairment. Theranostics 2020, 10, 7384–7400. [Google Scholar] [CrossRef] [PubMed]
- Shiba, T.; Makino, I.; Kawakami, K.; Kato, I.; Kobayashi, T.; Kaneko, K. p-Cresyl sulfate suppresses lipopolysaccharide-induced anti-bacterial immune responses in murine macrophages in vitro. Toxicol. Lett. 2016, 245, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Shiba, T.; Kawakami, K.; Sasaki, T.; Makino, I.; Kato, I.; Kobayashi, T.; Uchida, K.; Kaneko, K. Effects of intestinal bacteria-derived p-cresyl sulfate on Th1-type immune response in vivo and in vitro. Toxicol. Appl. Pharmacol. 2014, 274, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Parnaby, C.N.; Barrow, E.J.; Edirimanne, S.B.; Parrott, N.R.; Frizelle, F.A.; Watson, A.J.M. Colorectal complications of end-stage renal failure and renal transplantation: A review. Color. Dis. 2012, 14, 403–415. [Google Scholar] [CrossRef]
- Wu, I.-W.; Lin, C.-Y.; Chang, L.-C.; Lee, C.-C.; Chiu, C.-Y.; Hsu, H.-J.; Sun, C.-Y.; Chen, Y.-C.; Kuo, Y.-L.; Yang, C.-W.; et al. Gut Microbiota as Diagnostic Tools for Mirroring Disease Progression and Circulating Nephrotoxin Levels in Chronic Kidney Disease: Discovery and Validation Study. Int. J. Biol. Sci. 2020, 16, 420–434. [Google Scholar] [CrossRef]
- Liu, W.-C.; Tomino, Y.; Lu, K.-C. Impacts of Indoxyl Sulfate and p-Cresol Sulfate on Chronic Kidney Disease and Mitigating Effects of AST-120. Toxins 2018, 10, 367. [Google Scholar] [CrossRef]
- Dou, L.; Anfosso, F.; Sabatier, F.; Moal, V.; Glorieux, G.; De Smet, R.; Vanholder, R.; Dignat-George, F.; Sampol, J.; Berland, Y.; et al. p-cresol, a uremic retention solute, alters the endothelial barrier function in vitro. Thromb. Haemost. 2004, 92, 140–150. [Google Scholar] [CrossRef]
- Al Hinai, E.A.; Kullamethee, P.; Rowland, I.R.; Swann, J.; Walton, G.E.; Commane, D.M. Modelling the role of microbial p-cresol in colorectal genotoxicity. Gut Microbes 2018, 10, 398–411. [Google Scholar] [CrossRef]
- Peng, Y.-S.; Syu, J.-P.; Wang, S.-D.; Pan, P.-C.; Kung, H.-N. BSA-bounded p-cresyl sulfate potentiates the malignancy of bladder carcinoma by triggering cell migration and EMT through the ROS/Src/FAK signaling pathway. Cell Biol. Toxicol. 2019, 36, 287–300. [Google Scholar] [CrossRef]
- Wu, T.-K.; Wei, C.-W.; Pan, Y.-R.; Hsu, R.-J.; Wu, C.-Y.; Yu, Y.-L. The uremic toxin p-cresyl sulfate induces proliferation and migration of clear cell renal cell carcinoma via microRNA-21/HIF-1α axis signals. Sci. Rep. 2019, 9, 3207. [Google Scholar] [CrossRef]
- Yau, T.O.; Tang, C.-M.; Harriss, E.K.; Dickins, B.; Polytarchou, C. Faecal microRNAs as a non-invasive tool in the diagnosis of colonic adenomas and colorectal cancer: A meta-analysis. Sci. Rep. 2019, 9, 9491. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-H.; Huang, H.-P.; Chang, H.-R. The uremic toxin p-cresol promotes the invasion and migration on carcinoma cells via Ras and mTOR signaling. Toxicol. Vitr. 2019, 58, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.D.; Thapa, M.; Aminzadeh-Gohari, S.; Redtenbacher, A.-S.; Catalano, L.; Feichtinger, R.G.; Koelblinger, P.; Dallmann, G.; Emberger, M.; Kofler, B.; et al. Targeted Metabolomics Identifies Plasma Biomarkers in Mice with Metabolically Heterogeneous Melanoma Xenografts. Cancers 2021, 13, 434. [Google Scholar] [CrossRef]
- Valko-Rokytovská, M.; Hubková, B.; Birková, A.; Mašlanková, J.; Stupák, M.; Zábavníková, M.; Čižmárová, B.; Mareková, M. Specific Urinary Metabolites in Malignant Melanoma. Medicina 2019, 55, 145. [Google Scholar] [CrossRef]
- Rajasekaran, R.; Aruna, P.R.; Koteeswaran, D.; Bharanidharan, G.; Baludavid, M.; Ganesan, S. Steady-state and time-resolved fluorescence spectroscopic characterization of urine of healthy subjects and cervical cancer patients. J. Biomed. Opt. 2014, 19, 37003. [Google Scholar] [CrossRef]
- Kim, K.-B.; Yang, J.-Y.; Kwack, S.J.; Park, K.L.; Kim, H.S.; Ryu, D.H.; Kim, Y.-J.; Hwang, G.-S.; Lee, B.M. Toxicometabolomics of Urinary Biomarkers for Human Gastric Cancer in a Mouse Model. J. Toxicol. Environ. Health Part A 2010, 73, 1420–1430. [Google Scholar] [CrossRef]
- Chen, Z.; Huang, X.; Gao, Y.; Zeng, S.; Mao, W. Plasma-metabolite-based machine learning is a promising diagnostic approach for esophageal squamous cell carcinoma investigation. J. Pharm. Anal. 2020, 11, 505–514. [Google Scholar] [CrossRef]
- Rosenzweig, B.; Rubinstein, N.D.; Reznik, E.; Shingarev, R.; Juluru, K.; Akin, O.; Hsieh, J.J.; Jaimes, E.A.; Russo, P.; Susztak, K.; et al. Benign and tumor parenchyma metabolomic profiles affect compensatory renal growth in renal cell carcinoma surgical patients. PLoS ONE 2017, 12, e0180350. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.; Amaro, F.; Lima, A.R.; Carvalho-Maia, C.; Jerónimo, C.; Henrique, R.; Bastos, M.D.L.; Carvalho, M.; de Pinho, P.G. Urinary Volatilomics Unveils a Candidate Biomarker Panel for Noninvasive Detection of Clear Cell Renal Cell Carcinoma. J. Proteome Res. 2021, 20, 3068–3077. [Google Scholar] [CrossRef]
- Oto, J.; Fernández-Pardo, Á.; Roca, M.; Plana, E.; Solmoirago, M.J.; Sánchez-González, J.V.; Vera-Donoso, C.D.; Martínez-Sarmiento, M.; España, F.; Navarro, S.; et al. Urine metabolomic analysis in clear cell and papillary renal cell carcinoma: A pilot study. J. Proteom. 2020, 218, 103723. [Google Scholar] [CrossRef]
- Ravnik, Z.; Muthiah, I.; Dhanaraj, P. Computational studies on bacterial secondary metabolites against breast cancer. J. Biomol. Struct. Dyn. 2020, 39, 7056–7064. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-Y.; Chang, S.-C.; Wu, M.-S.; Casper, J.; Schmitz, J.; Bräsen, J.H.; Khalifa, A.; Schmidt, B.M.; Einecke, G.; Haller, H.; et al. Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int. 2012, 81, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Fandy, T.E. Development of DNA Methyltransferase Inhibitors for the Treatment of Neoplastic Diseases. Curr. Med. Chem. 2009, 16, 2075–2085. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Sawalha, A.H. Epigenetic regulation and the pathogenesis of systemic lupus erythematosus. Transl. Res. 2009, 153, 4–10. [Google Scholar] [CrossRef]
- Wolf, I.; Levanon-Cohen, S.; Bose, S.; Ligumsky, H.; Sredni, B.; Kanety, H.; Kuro-O, M.; Karlan, B.; Kaufman, B.; Koeffler, H.P.; et al. Klotho: A tumor suppressor and a modulator of the IGF-1 and FGF pathways in human breast cancer. Oncogene 2008, 27, 7094–7105. [Google Scholar] [CrossRef]
- Camilli, T.C.; Xu, M.; O’connell, M.P.; Chien, B.; Frank, B.P.; Subaran, S.; Indig, F.E.; Morin, P.J.; Hewitt, S.M.; Weeraratna, A.T. Loss of Klotho during melanoma progression leads to increased filamin cleavage, increased Wnt5A expression, and enhanced melanoma cell motility. Pigment. Cell Melanoma Res. 2010, 24, 175–186. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA Methylation and Cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef]
- Robert, M.-F.; Morin, S.; Beaulieu, N.; Gauthier, F.; Chute, I.C.; Barsalou, A.; MacLeod, A.R. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat. Genet. 2002, 33, 61–65. [Google Scholar] [CrossRef]
- Shimizu, H.; Bolati, D.; Adijiang, A.; Adelibieke, Y.; Muteliefu, G.; Enomoto, A.; Higashiyama, Y.; Higuchi, Y.; Nishijima, F.; Niwa, T. Indoxyl Sulfate Downregulates Renal Expression of Klotho through Production of ROS and Activation of Nuclear Factor-ĸB. Am. J. Nephrol. 2011, 33, 319–324. [Google Scholar] [CrossRef]
- Yang, K.; Nie, L.; Huang, Y.; Zhang, J.; Xiao, T.; Guan, X.; Zhao, J. Amelioration of uremic toxin indoxyl sulfate-induced endothelial cell dysfunction by Klotho protein. Toxicol. Lett. 2012, 215, 77–83. [Google Scholar] [CrossRef]
- Wang, L.; Gao, Z.; Wang, L.; Gao, Y. Upregulation of nuclear factor-κB activity mediates CYP24 expression and reactive oxygen species production in indoxyl sulfate-induced chronic kidney disease. Nephrology 2016, 21, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.-T.; Lin, S.-H. Uremic Toxins and Frailty in Patients with Chronic Kidney Disease: A Molecular Insight. Int. J. Mol. Sci. 2021, 22, 6270. [Google Scholar] [CrossRef] [PubMed]
- Tamada, S.; Asai, T.; Kuwabara, N.; Iwai, T.; Uchida, J.; Teramoto, K.; Kaneda, N.; Yukimura, T.; Komiya, T.; Nakatani, T.; et al. Molecular Mechanisms and Therapeutic Strategies of Chronic Renal Injury: The Role of Nuclear Factor κB Activation in the Development of Renal Fibrosis. J. Pharmacol. Sci. 2006, 100, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Stockler-Pinto, M.B.; Soulage, C.O.; Borges, N.A.; Cardozo, L.F.M.F.; Dolenga, C.J.; Nakao, L.S.; Pecoits-Filho, R.; Fouque, D.; Mafra, D. From bench to the hemodialysis clinic: Protein-bound uremic toxins modulate NF-κB/Nrf2 expression. Int. Urol. Nephrol. 2017, 50, 347–354. [Google Scholar] [CrossRef]
- Masai, N.; Tatebe, J.; Yoshino, G.; Morita, T. Indoxyl Sulfate Stimulates Monocyte Chemoattractant Protein-1 Expression in Human Umbilical Vein Endothelial Cells by Inducing Oxidative Stress through Activation of the NADPH Oxidase-Nuclear Factor-.KAPPA.B Pathway. Circ. J. 2010, 74, 2216–2224. [Google Scholar] [CrossRef]
- Motojima, M.; Hosokawa, A.; Yamato, H.; Muraki, T.; Yoshioka, T. Uremic toxins of organic anions up-regulate PAI-1 expression by induction of NF-κB and free radical in proximal tubular cells. Kidney Int. 2003, 63, 1671–1680. [Google Scholar] [CrossRef]
- Yang, J.; Li, H.; Zhang, C.; Zhou, Y. Indoxyl sulfate reduces Ito,f by activating ROS/MAPK and NF-κB signaling pathways. JCI Insight. 2022, 7, e145475. [Google Scholar] [CrossRef]
- Wakamatsu, T.; Yamamoto, S.; Ito, T.; Sato, Y.; Matsuo, K.; Takahashi, Y.; Kaneko, Y.; Goto, S.; Kazama, J.J.; Gejyo, F.; et al. Indoxyl Sulfate Promotes Macrophage IL-1β Production by Activating Aryl Hydrocarbon Receptor/NF-κ/MAPK Cascades, but the NLRP3 inflammasome Was not Activated. Toxins 2018, 10, 124. [Google Scholar] [CrossRef]
- Shimizu, H.; Yisireyili, M.; Higashiyama, Y.; Nishijima, F.; Niwa, T. Indoxyl sulfate upregulates renal expression of ICAM-1 via production of ROS and activation of NF-κB and p53 in proximal tubular cells. Life Sci. 2013, 92, 143–148. [Google Scholar] [CrossRef]
- Shimizu, H.; Bolati, D.; Higashiyama, Y.; Nishijima, F.; Shimizu, K.; Niwa, T. Indoxyl sulfate upregulates renal expression of MCP-1 via production of ROS and activation of NF-κB, p53, ERK, and JNK in proximal tubular cells. Life Sci. 2012, 90, 525–530. [Google Scholar] [CrossRef]
- Zubair, A.; Frieri, M. Role of Nuclear Factor-ĸB in Breast and Colorectal Cancer. Curr. Allergy Asthma Rep. 2012, 13, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Sheflin, A.; Whitney, A.; Weir, T.L. Cancer-Promoting Effects of Microbial Dysbiosis. Curr. Oncol. Rep. 2014, 16, 406. [Google Scholar] [CrossRef] [PubMed]
- Aranha, M.M.; Borralho, P.M.; Ravasco, P.; Da Silva, I.B.M.; Correia, L.; Fernandes, A.; Camilo, M.E.; Rodrigues, C.M.P. NF-?B and apoptosis in colorectal tumourigenesis. Eur. J. Clin. Investig. 2007, 37, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Katsube, Y.; Tsujimoto, M.; Koide, H.; Ochiai, M.; Hojyo, A.; Ogawa, K.; Kambara, K.; Torii, N.; Shima, D.; Furukubo, T.; et al. Cooperative inhibitory effects of uremic toxins and other serum components on OATP1B1-mediated transport of SN-38. Cancer Chemother. Pharmacol. 2017, 79, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, H.; Tsujimoto, M.; Shinmoto, T.; Ogino, H.; Oda, T.; Yoshida, T.; Furukubo, T.; Izumi, S.; Yamakawa, T.; Tachiki, H.; et al. Uremic Toxins Enhance Statin-Induced Cytotoxicity in Differentiated Human Rhabdomyosarcoma Cells. Toxins 2014, 6, 2612–2625. [Google Scholar] [CrossRef]
- Park, R.; Madhavaram, S.; Ji, J.D. The Role of Aryl-Hydrocarbon Receptor (AhR) in Osteoclast Differentiation and Function. Cells 2020, 9, 2294. [Google Scholar] [CrossRef]
- Sári, Z.; Mikó, E.; Kovács, T.; Boratkó, A.; Ujlaki, G.; Jankó, L.; Kiss, B.; Uray, K.; Bai, P. Indoxylsulfate, a Metabolite of the Microbiome, Has Cytostatic Effects in Breast Cancer via Activation of AHR and PXR Receptors and Induction of Oxidative Stress. Cancers 2020, 12, 2915. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, P.-P.; Fang, X.-Q.; Liu, Y.; Wang, J.-Q.; Zhou, H.-Z.; Chen, S.-T.; Chao, H. A ruthenium(II) complex containing a p-cresol group induces apoptosis in human cervical carcinoma cells through endoplasmic reticulum stress and reactive oxygen species production. J. Inorg. Biochem. 2018, 191, 126–134. [Google Scholar] [CrossRef]
- Belghasem, M.; Roth, D.; Richards, S.; Napolene, M.A.; Walker, J.; Yin, W.; Arinze, N.; Lyle, C.; Spencer, C.; Francis, J.M.; et al. Metabolites in a mouse cancer model enhance venous thrombogenicity through the aryl hydrocarbon receptor–tissue factor axis. Blood 2019, 134, 2399–2413. [Google Scholar] [CrossRef]
- Fourdinier, O.; Schepers, E.; Meuth, V.M.-L.; Glorieux, G.; Liabeuf, S.; Verbeke, F.; Vanholder, R.; Brigant, B.; Pletinck, A.; Diouf, M.; et al. Serum levels of miR-126 and miR-223 and outcomes in chronic kidney disease patients. Sci. Rep. 2019, 9, 4477. [Google Scholar] [CrossRef]
- Fourdinier, O.; Glorieux, G.; Brigant, B.; Diouf, M.; Pletinck, A.; Vanholder, R.; Choukroun, G.; Verbeke, F.; Massy, Z.A.; Meuth, V.M.-L.; et al. Syndecan-1 and Free Indoxyl Sulfate Levels Are Associated with miR-126 in Chronic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 10549. [Google Scholar] [CrossRef] [PubMed]
- Mármol, I.; Sánchez-De-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Youssef, O.; Lahti, L.; Kokkola, A.; Karla, T.; Tikkanen, M.; Ehsan, H.; Carpelan-Holmström, M.; Koskensalo, S.; Böhling, T.; Rautelin, H.; et al. Stool Microbiota Composition Differs in Patients with Stomach, Colon, and Rectal Neoplasms. Dig. Dis. Sci. 2018, 63, 2950–2958. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, G.; Qiu, Y.; Fei, N.; Zhang, M.; Pang, X.; Jia, W.; Cai, S.; Zhao, L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2011, 6, 320–329. [Google Scholar] [CrossRef]
- Rysz, J.; Franczyk, B.; Ławiński, J.; Olszewski, R.; Ciałkowska-Rysz, A.; Gluba-Brzózka, A. The Impact of CKD on Uremic Toxins and Gut Microbiota. Toxins 2021, 13, 252. [Google Scholar] [CrossRef]
- Simeoni, M.; Citraro, M.L.; Cerantonio, A.; Deodato, F.; Provenzano, M.; Cianfrone, P.; Capria, M.; Corrado, S.; Libri, E.; Comi, A.; et al. An open-label, randomized, placebo-controlled study on the effectiveness of a novel probiotics administration protocol (ProbiotiCKD) in patients with mild renal insufficiency (stage 3a of CKD). Eur. J. Nutr. 2018, 58, 2145–2156. [Google Scholar] [CrossRef]
- Li, F.; Yang, X.-W.; Krausz, K.W.; Nichols, R.G.; Xu, W.; Patterson, A.D.; Gonzalez, F.J. Modulation of Colon Cancer by Nutmeg. J. Proteome Res. 2015, 14, 1937–1946. [Google Scholar] [CrossRef]
- Adesso, S.; Popolo, A.; Bianco, G.; Sorrentino, R.; Pinto, A.; Autore, G.; Marzocco, S. The Uremic Toxin Indoxyl Sulphate Enhances Macrophage Response to LPS. PLoS ONE 2013, 8, e76778. [Google Scholar] [CrossRef]
- Hauser, A.B.; Stinghen, A.E.M.; Kato, S.; Bucharles, S.; Aita, C.; Yuzawa, Y.; Pecoits-Filho, R. Characteristics and causes of immune dysfunction related to uremia and dialysis. Perit. Dial. Int. 2008, 28 (Suppl. S3), S183–S187. [Google Scholar] [CrossRef]
- Shiba, T.; Makino, I.; Sasaki, T.; Fukuhara, Y.; Kawakami, K.; Kato, I.; Kobayashi, T. p-Cresyl sulfate decreases peripheral B cells in mice with adenine-induced renal dysfunction. Toxicol. Appl. Pharmacol. 2018, 342, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.-L.; Shu, K.-H.; Yang, F.-J.; Chou, T.-Y.; Chen, P.-M.; Lay, F.-Y.; Pan, S.-Y.; Lin, C.-J.; Litjens, N.H.R.; Betjes, M.G.H.; et al. A comprehensive characterization of aggravated aging-related changes in T lymphocytes and monocytes in end-stage renal disease: The iESRD study. Immun. Ageing 2018, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Bonan, N.B.; Schepers, E.; Pecoits-Filho, R.; Dhondt, A.; Pletinck, A.; De Somer, F.; Vanholder, R.; Van Biesen, W.; Moreno-Amaral, A.; Glorieux, G. Contribution of the uremic milieu to an increased pro-inflammatory monocytic phenotype in chronic kidney disease. Sci. Rep. 2019, 9, 10236. [Google Scholar] [CrossRef] [PubMed]
- Tungsanga, S.; Panpetch, W.; Bhunyakarnjanarat, T.; Udompornpitak, K.; Katavetin, P.; Chancharoenthana, W.; Chatthanathon, P.; Somboonna, N.; Tungsanga, K.; Tumwasorn, S.; et al. Uremia-Induced Gut Barrier Defect in 5/6 Nephrectomized Mice Is Worsened by Candida Administration through a Synergy of Uremic Toxin, Lipopolysaccharide, and (1→3)-β-D-Glucan, but Is Attenuated by Lacticaseibacillus rhamnosus L34. Int. J. Mol. Sci. 2022, 23, 2511. [Google Scholar] [CrossRef] [PubMed]
- Carini, F.; Mazzola, M.; Rappa, F.; Jurjus, A.; Geagea, A.G.; Al Kattar, S.; Bou-Assi, T.; Jurjus, R.; Damiani, P.; Leone, A.; et al. Colorectal Carcinogenesis: Role of Oxidative Stress and Antioxidants. Anticancer. Res. 2017, 37, 4759–4766. [Google Scholar] [CrossRef] [PubMed]
- Adesso, S.; Ruocco, M.; Rapa, S.F.; Dal Piaz, F.; Di Iorio, B.R.; Popolo, A.; Autore, G.; Nishijima, F.; Pinto, A.; Marzocco, S. Effect of Indoxyl Sulfate on the Repair and Intactness of Intestinal Epithelial Cells: Role of Reactive Oxygen Species’ Release. Int. J. Mol. Sci. 2019, 20, 2280. [Google Scholar] [CrossRef]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Lau, W.L.; Liu, S.-M.; Pahlevan, S.; Yuan, J.; Khazaeli, M.; Ni, Z.; Chan, J.Y.; Vaziri, N.D. Role of Nrf2 Dysfunction in Uremia-Associated Intestinal Inflammation and Epithelial Barrier Disruption. Dig. Dis. Sci. 2015, 60, 1215–1222. [Google Scholar] [CrossRef]
- Stockler-Pinto, M.B.; Fouque, D.; Soulage, C.O.; Croze, M.; Mafra, D. Indoxyl Sulfate and p-Cresyl Sulfate in Chronic Kidney Disease. Could These Toxins Modulate the Antioxidant Nrf2-Keap1 Pathway? J. Ren. Nutr. 2014, 24, 286–291. [Google Scholar] [CrossRef]
- Triantafillidis, J.K.; Nasioulas, G.; Kosmidis, P.A. Colorectal cancer and inflammatory bowel disease: Epidemiology, risk factors, mechanisms of carcinogenesis and prevention strategies. Anticancer. Res. 2009, 29, 2727–2737. [Google Scholar]
- Andriamihaja, M.; Lan, A.; Beaumont, M.; Audebert, M.; Wong, X.; Yamada, K.; Yin, Y.; Tomé, D.; Carrasco-Pozo, C.; Gotteland, M.; et al. The deleterious metabolic and genotoxic effects of the bacterial metabolite p-cresol on colonic epithelial cells. Free. Radic. Biol. Med. 2015, 85, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Li, C.; Chen, W.; Zeng, Y.; Yang, H.; Hu, Y.; Song, H.; Zeng, X.; Li, Q.; Fu, P. Causal Association between Chronic Kidney Disease and Risk of 19 Site-Specific Cancers: A Mendelian Randomization Study. Cancer Epidemiol Biomark. Prev. 2022, 31, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.S.; Elyan, B.M.P.; Herrmann, S.M.; Lang, N.N.; Jones, R.J.; Mark, P.B. The ‘other’ big complication: How chronic kidney disease impacts on cancer risks and outcomes. Nephrol. Dial. Transplant. 2022, 38, 1071–1079. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| MICROBIOTA | CKD | CRC |
|---|---|---|
| +Enterobacteriaceae | Responsible to produce p-Cresol (p-C) [8] | Responsible for the development of chronic intestinal inflammation which favors tumor development [63] |
| +Escherichia coli | Mainly responsible for the formation of indoxyl sulfate (IS) [8] | -Promotes tumorigenesis -Down-regulates DNA mismatch repair proteins [64,65] |
| +Ruminococcus | Responsible for the production of p-Cresyl Sulfate (p-CS) [8] | Patients with a high risk of CRC have significantly higher levels of these bacteria [65] |
| +Bacteroides | Capable of fermenting aromatic amino acids to produce potentially bioactive products, including phenols, indoles and p-C [61] | Possess the ability to exert carcinogenic effects through the alkylation of DNA [63] |
| −Lactobacillaceae | Fundamental role in maintaining gastrointestinal homeostasis and preventing the development of cancer and the migration of cancer cells [8,64,66,67] | |
| −Bifidobacteriaceae | Contribute to de novo biosynthesis of folate in the intestine, and their deficiency can cause chromosomal instability and increased risk of aneuploidy associated with rectal cancer [64] | |
| Toxins | References | Role in Cancer | Study Design | Study Group |
|---|---|---|---|---|
| p-CRESIL SULFATE (p-CS) | Shiba T et al. [12] | ↓macrophage activity | Experimental | Cells: RAW264.7 (macrophage-like cell line) |
| Shiba T et al. [13] | ↓Th1-type immune response | Experimental | Mouse model: female BALB/c mice cells: splenocyte cultures | |
| Peng YS et al. [19] | ↑EMT ↑ROS | Experimental | Cells: TSGH-8301 (bladder cancer cells) | |
| Wu TK et al. [20] | ↑EMT ↑HIF-1α ↑miR-21 | Experimental | Cells: A498 and 786-O (human CRcc cells) | |
| Li F et al. [68] | ↑IL-6 | Experimental | Cells: Caco2 (normal colon cell line) | |
| Shiba T et al. [71] | ↓peripheral B lymphocytes | Experimental | Mouse model with renal dysfunction | |
| Chiu YL et al. [72] | ↑CD8+ T cells | Cross-sectional | 412 patients with end-stage renal disease | |
| Bonan BN et al. [73] | ↑CD14 ++ ↑CD16 + | Experimental | 220 patients | |
| INDOXYL SULFATE (IS) | Rapa SF et al. [10] | ↑intestinal ROS production ↑SOD2 ↑HO1 ↑intestinal expression of COX 2 ↑iNOS ↑TNF-α ↑IL-6 ↑IL-1β | Experimental | Mouse model: C57BL/6J Cells: IEC-6(normal small intestine cell line) |
| Huang Y et al. [11] | ↑macroscopic intestinal damage ↑ pro-inflammatory cytokines ↑intestinal ROS production ↑IRF1 ↓DRP1 | Experimental | Mouse models: IRF1 knockout, BALB/c with CKD Cells: Caco2 (normal colon cell line) | |
| Li F et al. [68] | ↑IL-6 | Experimental | Cells: Caco2 (normal colon cell line) | |
| Adesso S et al. [69] | ↑(NO), ↑iNOS ↑COX-2 ↑TNF-α ↑IL-6 | Experimental | Cells: macrophages J774A | |
| Bonan BN et al. [73] | ↑CD14 ++ ↑CD16 + | Experimental | 220 patients | |
| Tungsanga S et al. [74] | ↓cells vitality ↓transepithelial electrical resistance | Experimental | Cells: Caco2 (normal colon cell line) | |
| Adesso S et al. [76] | ↑intestinal ROS | Experimental | Cells: IEC-6(normal small intestine cell line) | |
| Dou L et al. [77] | ↑ROS ↑NAD (P) H activity ↓Glutathione | Experimental | Cells: HUVEC (endothelial cell line) | |
| p-Cresol (p-C) | Hsu YH et al. [22] | ↑genotoxicity | Experimental | Cells: HT29 (colon cancer cell line) Caco2(normal colon cell line) IEC-6(normal small intestine cell line) |
| Hinai EAA et al. [18] Andriamihaja M et al. [81] | ↑genotoxicity ↓ATP | Experimental | Cells: HT29-Glc−/+ (colon cancer cell line) IEC-6(normal small intestine cell line) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Paola, R.; De, A.; Izhar, R.; Abate, M.; Zappavigna, S.; Capasso, A.; Perna, A.F.; La Russa, A.; Capasso, G.; Caraglia, M.; et al. Possible Effects of Uremic Toxins p-Cresol, Indoxyl Sulfate, p-Cresyl Sulfate on the Development and Progression of Colon Cancer in Patients with Chronic Renal Failure. Genes 2023, 14, 1257. https://doi.org/10.3390/genes14061257
Di Paola R, De A, Izhar R, Abate M, Zappavigna S, Capasso A, Perna AF, La Russa A, Capasso G, Caraglia M, et al. Possible Effects of Uremic Toxins p-Cresol, Indoxyl Sulfate, p-Cresyl Sulfate on the Development and Progression of Colon Cancer in Patients with Chronic Renal Failure. Genes. 2023; 14(6):1257. https://doi.org/10.3390/genes14061257
Chicago/Turabian StyleDi Paola, Rossella, Ananya De, Raafiah Izhar, Marianna Abate, Silvia Zappavigna, Anna Capasso, Alessandra F. Perna, Antonella La Russa, Giovambattista Capasso, Michele Caraglia, and et al. 2023. "Possible Effects of Uremic Toxins p-Cresol, Indoxyl Sulfate, p-Cresyl Sulfate on the Development and Progression of Colon Cancer in Patients with Chronic Renal Failure" Genes 14, no. 6: 1257. https://doi.org/10.3390/genes14061257
APA StyleDi Paola, R., De, A., Izhar, R., Abate, M., Zappavigna, S., Capasso, A., Perna, A. F., La Russa, A., Capasso, G., Caraglia, M., & Simeoni, M. (2023). Possible Effects of Uremic Toxins p-Cresol, Indoxyl Sulfate, p-Cresyl Sulfate on the Development and Progression of Colon Cancer in Patients with Chronic Renal Failure. Genes, 14(6), 1257. https://doi.org/10.3390/genes14061257