
Prediction of Tumor Development and Urine-Based Liquid Biopsy for Molecule-Targeted Therapy of Gliomas
Abstract
Simple Summary
Abstract
1. Introduction
2. Mathematical Model of Tumor Development
3. Urine-Based Genetic–Molecular Diagnosis
4. Molecule-Targeted Therapy
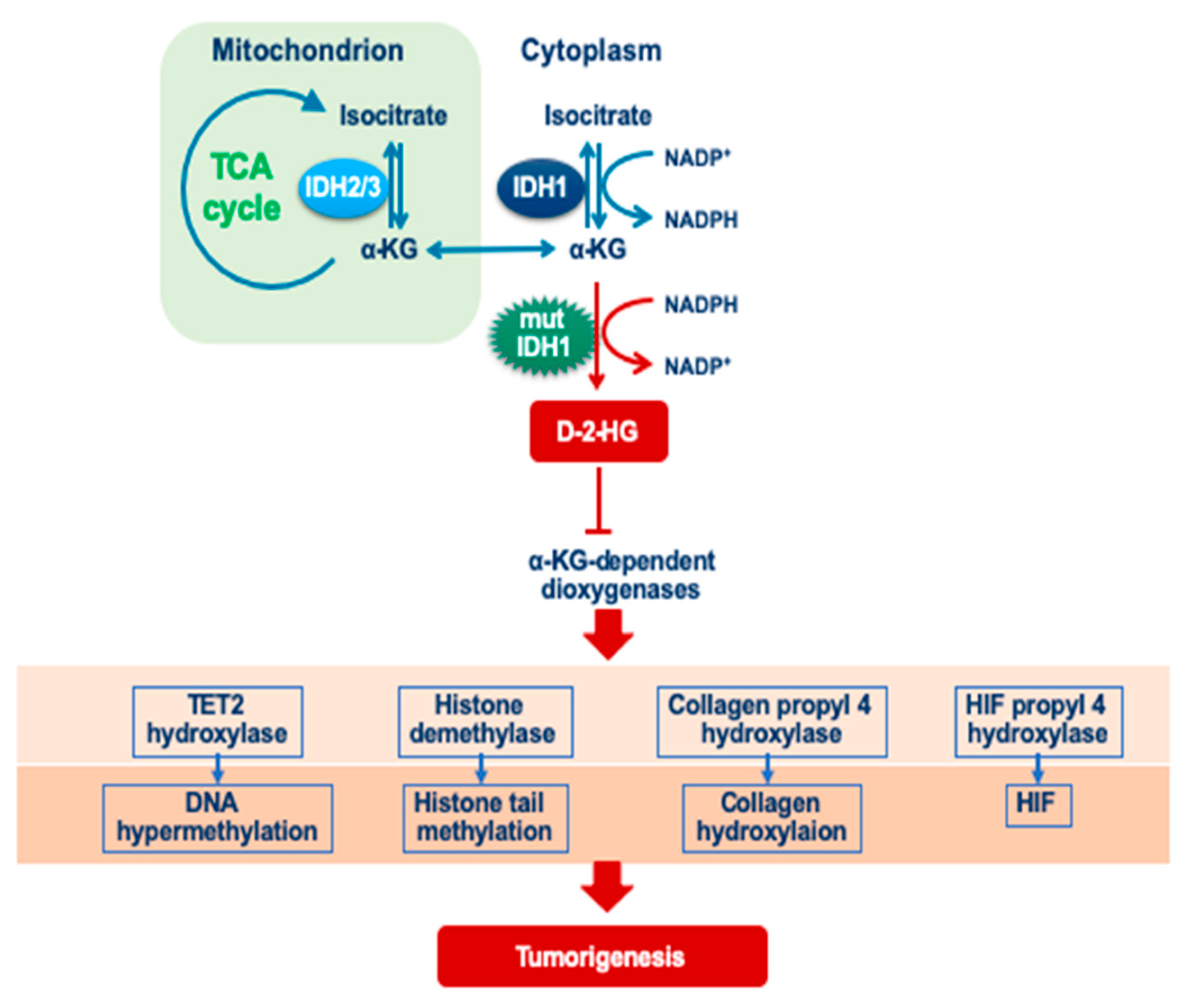
4.1. Isocitrate Dehydrogenase
4.2. TERT
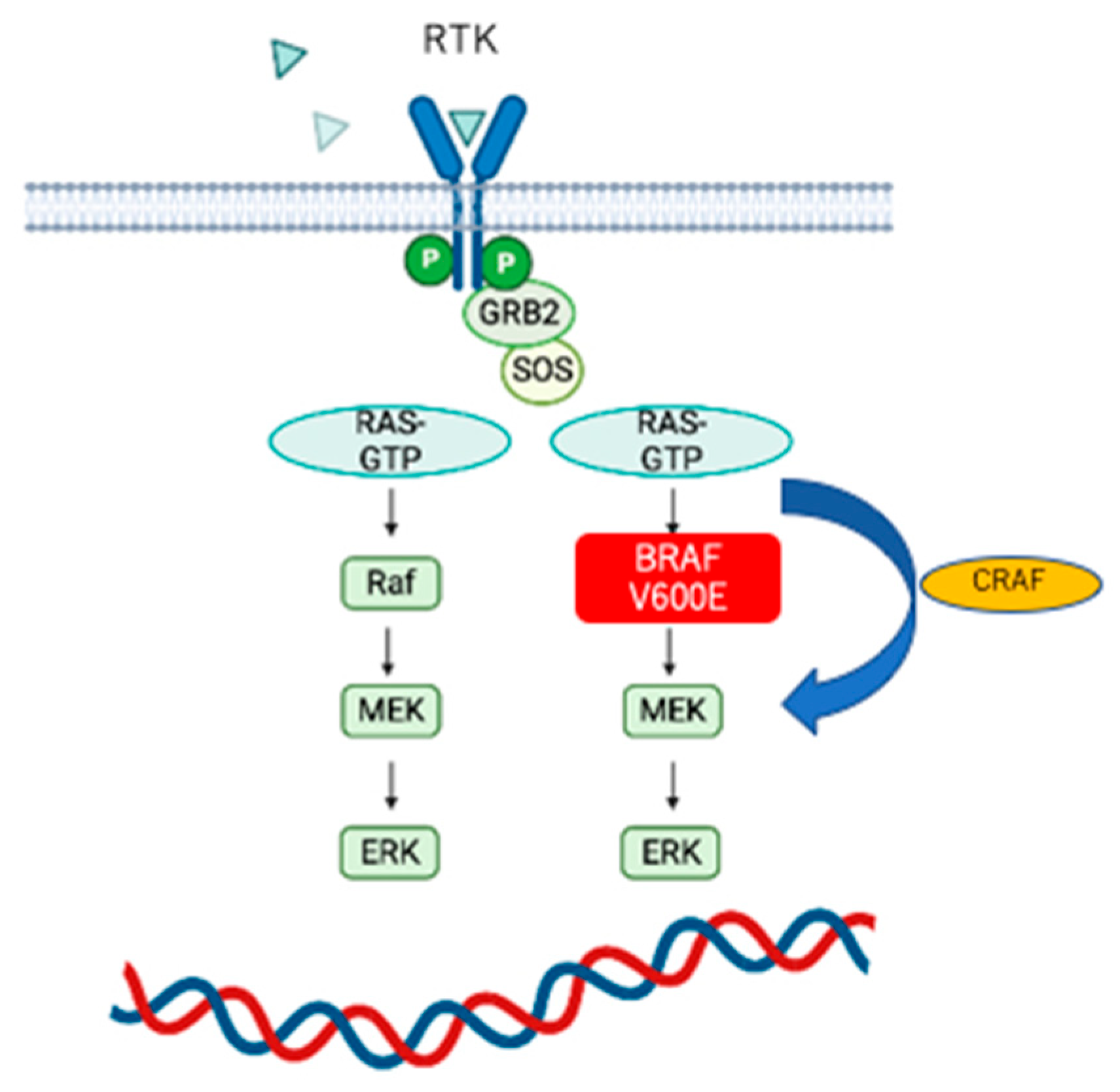
4.3. BRAF
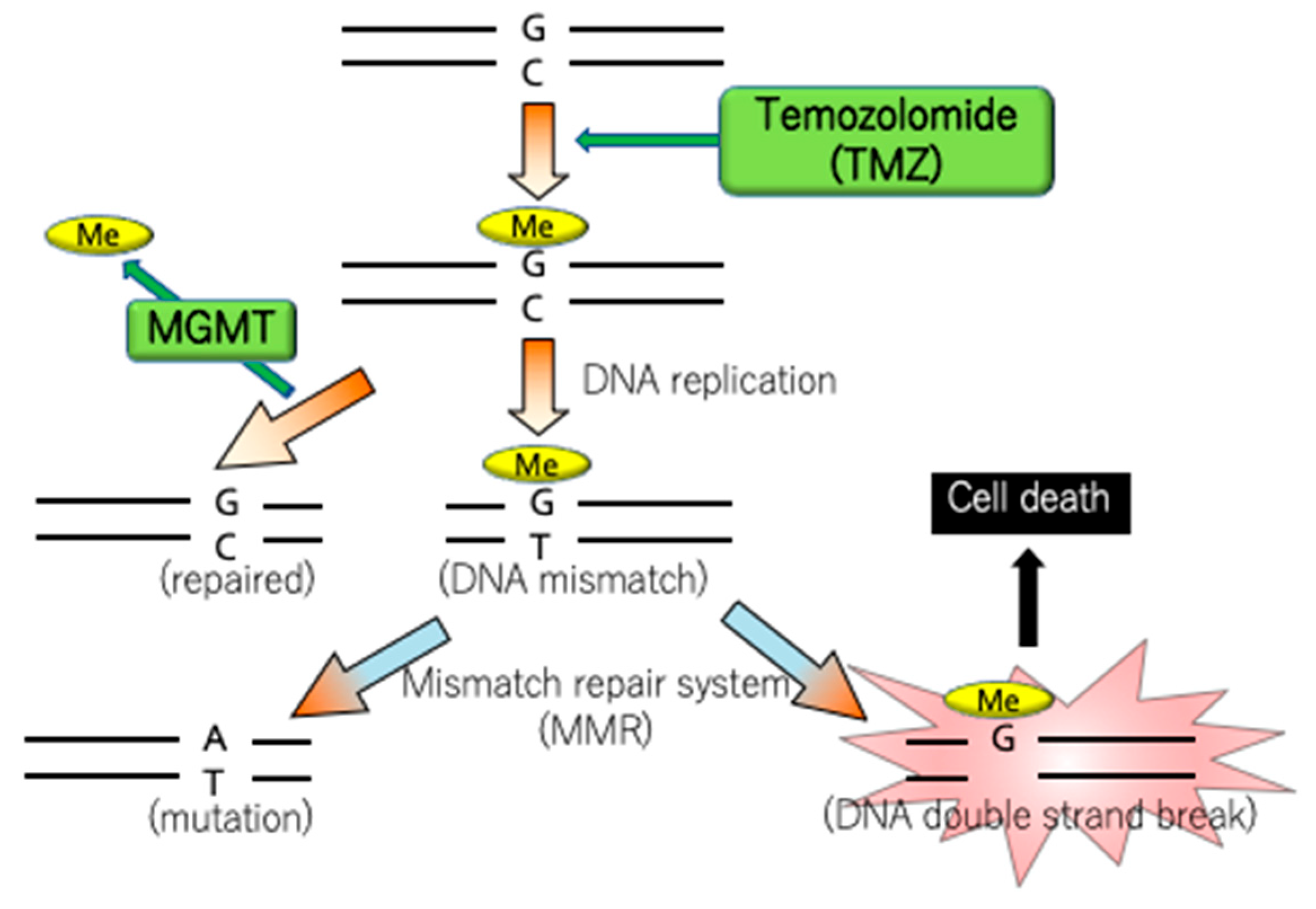
4.4. MGMT
4.5. H3K27/H3G34
4.6. NTRK-Fusion and ROS1-Fusion Genes
5. Discussion
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Rouse, C.; Chen, Y.; Dowling, J.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro-Oncol. 2014, 16 (Suppl. S4), iv1–iv63. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Fric, R.; Eide, P.K. Comparative observational study on the clinical presentation, intracranial volume measurements, and intracranial pressure scores in patients with either Chiari malformation Type I or idiopathic intracranial hypertension. J. Neurosurg. 2016, 126, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N. WHO Classification of Tumours of the Central Nervous System; International Agency for Research on Cancer: Lyon, France, 2016.
- Garcia, D.M.; Fulling, K.H.; Marks, J.E. The value of radiation therapy in addition to surgery for astrocytomas of the adult cerebrum. Cancer 1985, 55, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Karim, A.B.; Maat, B.; Hatlevoll, R.; Menten, J.; Rutten, E.H.; Thomas, D.G.; Mascarenhas, F.; Horiot, J.C.; Parvinen, L.M.; van Reijn, M.; et al. A randomized trial on dose-response in radiation therapy of low-grade cerebral glioma: European Organization for Research and Treatment of Cancer (EORTC) Study 22844. Int. J. Radiat. Oncol. Biol. Phys. 1996, 36, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Buckner, J.C.; Shaw, E.G.; Pugh, S.L.; Chakravarti, A.; Gilbert, M.R.; Barger, G.R.; Coons, S.; Ricci, P.; Bullard, D.; Brown, P.D.; et al. Radiation plus Procarbazine, CCNU, and Vincristine in Low-Grade Glioma. N. Engl. J. Med. 2016, 374, 1344–1355. [Google Scholar] [CrossRef]
- van den Bent, M.J.; Taphoorn, M.J.; Brandes, A.A.; Menten, J.; Stupp, R.; Frenay, M.; Chinot, O.; Kros, J.M.; van der Rijt, C.C.; Vecht, C.J.; et al. Phase II study of first-line chemotherapy with temozolomide in recurrent oligodendroglial tumors: The European Organization for Research and Treatment of Cancer Brain Tumor Group Study 26971. J. Clin. Oncol. 2003, 21, 2525–2528. [Google Scholar] [CrossRef]
- Cairncross, J.G.; Ueki, K.; Zlatescu, M.C.; Lisle, D.K.; Finkelstein, D.M.; Hammond, R.R.; Silver, J.S.; Stark, P.C.; Macdonald, D.R.; Ino, Y.; et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J. Natl. Cancer Inst. 1998, 90, 1473–1479. [Google Scholar] [CrossRef]
- Fisher, B.J.; Pugh, S.L.; Macdonald, D.R.; Chakravatri, A.; Lesser, G.J.; Fox, S.; Rogers, C.L.; Werner-Wasik, M.; Doyle, T.; Bahary, J.P.; et al. Phase 2 Study of a Temozolomide-Based Chemoradiation Therapy Regimen for High-Risk, Low-Grade Gliomas: Long-Term Results of Radiation Therapy Oncology Group 0424. Int. J. Radiat Oncol. Biol. Phys. 2020, 107, 720–725. [Google Scholar] [CrossRef]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015, 47, 458–468. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef]
- Smith, J.S.; Perry, A.; Borell, T.J.; Lee, H.K.; O’Fallon, J.; Hosek, S.M.; Kimmel, D.; Yates, A.; Burger, P.C.; Scheithauer, B.W.; et al. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J. Clin. Oncol. 2000, 18, 636–645. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. Genetic profile of astrocytic and oligodendroglial gliomas. Brain Tumor Pathol. 2011, 28, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Balss, J.; Meyer, J.; Mueller, W.; Korshunov, A.; Hartmann, C.; von Deimling, A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008, 116, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Meyer, J.; Balss, J.; Capper, D.; Mueller, W.; Christians, A.; Felsberg, J.; Wolter, M.; Mawrin, C.; Wick, W.; et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: A study of 1010 diffuse gliomas. Acta Neuropathol. 2009, 118, 469–474. [Google Scholar] [CrossRef]
- Bai, H.; Harmanci, A.S.; Erson-Omay, E.Z.; Li, J.; Coskun, S.; Simon, M.; Krischek, B.; Ozduman, K.; Omay, S.B.; Sorensen, E.A.; et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat. Genet. 2016, 48, 59–66. [Google Scholar] [CrossRef]
- de Souza, C.F.; Sabedot, T.S.; Malta, T.M.; Stetson, L.; Morozova, O.; Sokolov, A.; Laird, P.W.; Wiznerowicz, M.; Iavarone, A.; Snyder, J.; et al. A Distinct DNA Methylation Shift in a Subset of Glioma CpG Island Methylator Phenotypes during Tumor Recurrence. Cell Rep. 2018, 23, 637–651. [Google Scholar] [CrossRef]
- Bogdanska, M.U.; Bodnar, M.; Piotrowska, M.J.; Murek, M.; Schucht, P.; Beck, J.; Martinez-Gonzalez, A.; Perez-Garcia, V.M. A mathematical model describes the malignant transformation of low grade gliomas: Prognostic implications. PLoS ONE 2017, 12, e0179999. [Google Scholar] [CrossRef]
- Mandonnet, E.; Delattre, J.Y.; Tanguy, M.L.; Swanson, K.R.; Carpentier, A.F.; Duffau, H.; Cornu, P.; Van Effenterre, R.; Alvord, E.C.; Capelle, L. Continuous growth of mean tumor diameter in a subset of grade II gliomas. Ann. Neurol. 2003, 53, 524–528. [Google Scholar] [CrossRef]
- Aoki, K.; Suzuki, H.; Yamamoto, T.; Yamamoto, K.N.; Maeda, S.; Okuno, Y.; Ranjit, M.; Motomura, K.; Ohka, F.; Tanahashi, K.; et al. Mathematical Modeling and Mutational Analysis Reveal Optimal Therapy to Prevent Malignant Transformation in Grade II IDH-Mutant Gliomas. Cancer Res. 2021, 81, 4861–4873. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 2019, 25, 1415–1421. [Google Scholar] [CrossRef]
- Thierry, A.R.; Mouliere, F.; El Messaoudi, S.; Mollevi, C.; Lopez-Crapez, E.; Rolet, F.; Gillet, B.; Gongora, C.; Dechelotte, P.; Robert, B.; et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat. Med. 2014, 20, 430–435. [Google Scholar] [CrossRef]
- Zill, O.A.; Banks, K.C.; Fairclough, S.R.; Mortimer, S.A.; Vowles, J.V.; Mokhtari, R.; Gandara, D.R.; Mack, P.C.; Odegaard, J.I.; Nagy, R.J.; et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin. Cancer. Res. 2018, 24, 3528–3538. [Google Scholar] [CrossRef]
- Sheridan, C. Investors keep the faith in cancer liquid biopsies. Nat. Biotechnol. 2019, 37, 972–974. [Google Scholar] [CrossRef]
- Yasui, T.; Paisrisarn, P.; Yanagida, T.; Konakade, Y.; Nakamura, Y.; Nagashima, K.; Musa, M.; Thiodorus, I.A.; Takahashi, H.; Naganawa, T.; et al. Molecular profiling of extracellular vesicles via charge-based capture using oxide nanowire microfluidics. Biosens. Bioelectron. 2021, 194, 113589. [Google Scholar] [CrossRef]
- Yasui, T.; Yanagida, T.; Ito, S.; Konakade, Y.; Takeshita, D.; Naganawa, T.; Nagashima, K.; Shimada, T.; Kaji, N.; Nakamura, Y.; et al. Unveiling massive numbers of cancer-related urinary-microRNA candidates via nanowires. Sci. Adv. 2017, 3, e1701133. [Google Scholar] [CrossRef]
- Paisrisarn, P.; Yasui, T.; Zhu, Z.T.; Klamchuen, A.; Kasamechonchung, P.; Wutikhun, T.; Yordsri, V.; Baba, Y. Tailoring ZnO nanowire crystallinity and morphology for label-free capturing of extracellular vesicles. Nanoscale 2022, 14, 4484–4494. [Google Scholar] [CrossRef] [PubMed]
- Kitano, Y.; Aoki, K.; Ohka, F.; Yamazaki, S.; Motomura, K.; Tanahashi, K.; Hirano, M.; Naganawa, T.; Iida, M.; Shiraki, Y.; et al. Urinary MicroRNA-Based Diagnostic Model for Central Nervous System Tumors Using Nanowire Scaffolds. ACS Appl. Mater. Interfaces 2021, 13, 17316–17329. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Sasaki, M.; Knobbe, C.B.; Itsumi, M.; Elia, A.J.; Harris, I.S.; Chio, I.I.C.; Cairns, R.A.; McCracken, S.; Wakeham, A.; Haight, J.; et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012, 26, 2038–2049. [Google Scholar] [CrossRef]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef]
- Dang, L.; Su, S.M. Isocitrate Dehydrogenase Mutation and (R)-2-Hydroxyglutarate: From Basic Discovery to Therapeutics Development. Annu. Rev. Biochem. 2017, 86, 305–331. [Google Scholar] [CrossRef]
- Boddu, P.; Borthakur, G. Therapeutic targeting of isocitrate dehydrogenase mutant AML. Expert Opin. Investig. Drugs 2017, 26, 525–530. [Google Scholar] [CrossRef]
- Natsume, A.; Arakawa, Y.; Narita, Y.; Sugiyama, K.; Hata, N.; Muragaki, Y.; Shinojima, N.; Kumabe, T.; Saito, R.; Motomura, K.; et al. The first-in-human phase I study of a brain-penetrant mutant IDH1 inhibitor DS-1001 in patients with recurrent or progressive IDH1-mutant gliomas. Neuro Oncol. 2023, 25, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Chiba, K.; Lorbeer, F.K.; Shain, A.H.; McSwiggen, D.T.; Schruf, E.; Oh, A.; Ryu, J.; Darzacq, X.; Bastian, B.C.; Hockemeyer, D. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science 2017, 357, 1416–1420. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Miki, S.; Fujimoto, K.; Fukuoka, K.; Matsushita, Y.; Maida, Y.; Yasukawa, M.; Hayashi, M.; Shinkyo, R.; Kikuchi, K.; et al. Eribulin penetrates brain tumor tissue and prolongs survival of mice harboring intracerebral glioblastoma xenografts. Cancer Sci. 2019, 110, 2247–2257. [Google Scholar] [CrossRef]
- Liu, R.; Tan, J.; Shen, X.; Jiang, K.; Wang, C.; Zhu, G.; Xing, M. Therapeutic targeting of FOS in mutant TERT cancers through removing TERT suppression of apoptosis via regulating survivin and TRAIL-R2. Proc. Natl. Acad. Sci. USA 2021, 118, e2022779118. [Google Scholar] [CrossRef] [PubMed]
- Maida, Y.; Yasukawa, M.; Okamoto, N.; Ohka, S.; Kinoshita, K.; Totoki, Y.; Ito, T.K.; Minamino, T.; Nakamura, H.; Yamaguchi, S.; et al. Involvement of telomerase reverse transcriptase in heterochromatin maintenance. Mol. Cell Biol. 2014, 34, 1576–1593. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.J.; Rube, H.T.; Kreig, A.; Mancini, A.; Fouse, S.D.; Nagarajan, R.P.; Choi, S.; Hong, C.; He, D.; Pekmezci, M.; et al. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 2015, 348, 1036–1039. [Google Scholar] [CrossRef]
- Liu, R.; Zhang, T.; Zhu, G.; Xing, M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat. Commun. 2018, 9, 579. [Google Scholar] [CrossRef] [PubMed]
- Nobre, L.; Zapotocky, M.; Ramaswamy, V.; Ryall, S.; Bennett, J.; Alderete, D.; Balaguer Guill, J.; Baroni, L.; Bartels, U.; Bavle, A.; et al. Outcomes of BRAF V600E Pediatric Gliomas Treated With Targeted BRAF Inhibition. JCO Precis. Oncol. 2020, 4, 561–571. [Google Scholar] [CrossRef]
- Wen, P.Y.; Stein, A.; van den Bent, M.; De Greve, J.; Wick, A.; de Vos, F.Y.F.L.; von Bubnoff, N.; van Linde, M.E.; Lai, A.; Prager, G.W.; et al. Dabrafenib plus trametinib in patients with BRAFV600E-mutant low-grade and high-grade glioma (ROAR): A multicentre, open-label, single-arm, phase 2, basket trial. Lancet Oncol. 2022, 23, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Saborowski, A.; Yue, J.; Solomon, M.; Joseph, E.; Gadal, S.; Saborowski, M.; Kastenhuber, E.; Fellmann, C.; Ohara, K.; et al. Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell 2014, 25, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Ohka, F.; Natsume, A.; Motomura, K.; Kishida, Y.; Kondo, Y.; Abe, T.; Nakasu, Y.; Namba, H.; Wakai, K.; Fukui, T.; et al. The Global DNA Methylation Surrogate LINE-1 Methylation Is Correlated with MGMT Promoter Methylation and Is a Better Prognostic Factor for Glioma. PLoS ONE 2011, 6, e23332. [Google Scholar] [CrossRef] [PubMed]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, T.; Natsume, A.; Mizusawa, J.; Katayama, H.; Fukuda, H.; Sumi, M.; Nishikawa, R.; Narita, Y.; Muragaki, Y.; Maruyama, T.; et al. JCOG0911 INTEGRA study: A randomized screening phase II trial of interferonβ plus temozolomide in comparison with temozolomide alone for newly diagnosed glioblastoma. J. Neurooncol. 2018, 138, 627–636. [Google Scholar] [CrossRef]
- Natsume, A.; Aoki, K.; Ohka, F.; Maeda, S.; Hirano, M.; Adilijiang, A.; Motomura, K.; Sumi, M.; Nishikawa, R.; Narita, Y.; et al. Genetic analysis in patients with newly diagnosed glioblastomas treated with interferon-beta plus temozolomide in comparison with temozolomide alone. J. Neurooncol. 2020, 148, 17–27. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Hashizume, R.; Andor, N.; Ihara, Y.; Lerner, R.; Gan, H.; Chen, X.; Fang, D.; Huang, X.; Tom, M.W.; Ngo, V.; et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 2014, 20, 1394–1396. [Google Scholar] [CrossRef]
- Keane, L.; Cheray, M.; Saidi, D.; Kirby, C.; Friess, L.; Gonzalez-Rodriguez, P.; Gerdes, M.E.; Grabert, K.; McColl, B.W.; Joseph, B. Inhibition of microglial EZH2 leads to anti-tumoral effects in pediatric diffuse midline gliomas. Neurooncol. Adv. 2021, 3, vdab096. [Google Scholar] [CrossRef]
- Charest, A.; Lane, K.; McMahon, K.; Park, J.; Preisinger, E.; Conroy, H.; Housman, D. Fusion of FIG to the receptor tyrosine kinase ROS in a glioblastoma with an interstitial del(6)(q21q21). Genes Chromosomes Cancer 2003, 37, 58–71. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Lin, S.F. The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro Stucklin, A.S.; Ryall, S.; Fukuoka, K.; Zapotocky, M.; Lassaletta, A.; Li, C.; Bridge, T.; Kim, B.; Arnoldo, A.; Kowalski, P.E.; et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat. Commun. 2019, 10, 4343. [Google Scholar] [CrossRef] [PubMed]
- Sievers, P.; Stichel, D.; Sill, M.; Schrimpf, D.; Sturm, D.; Selt, F.; Ecker, J.; Kazdal, D.; Miele, E.; Kranendonk, M.E.G.; et al. GOPC:ROS1 and other ROS1 fusions represent a rare but recurrent drug target in a variety of glioma types. Acta Neuropathol. 2021, 142, 1065–1069. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Integrated Diagnosis | Oligodendroglioma, IDH-Mutant and 1p/19q-Codeleted, WHO Grade 2 or 3 | Astrocytoma, IDH-Mutant, WHO Grade 2 or 3 | Astrocytoma, IDH-Mutant, WHO Grade 4 | Glioblastoma, IDH Wild-Type, WHO Grade 4 |
|---|---|---|---|---|
| IDH | Mutant | Mutant | Mutant | Wild-type |
| ATRX | Nuclear ATRX retained | Nuclear ATRX lost | Nuclear ATRX lost | Nuclear ATRX retained |
| 1p/19q | Codeleted | Intact | Intact | |
| CDKN2A/B | Retained | Homozygously deleted ** | ||
| TERT, EGFR and/or +7/−10 * | TERT-mutant | TERT-mutant, EGFR-amplified and/or +7/−10 *** | ||
| Morphology | Necrosis and/or MVP ** | Necrosis and/or MVP *** |
| Diagnosis | Diffuse Midline Glioma, H3 K27-Altered, WHO Grade 4 | Diffuse Hemispheric Glioma, H3 G34-Mutant, WHO Grade 4 |
|---|---|---|
| IDH | Wild type | Wild type |
| ATRX | Nuclear ATRX retained | Nuclear ATRX > retained |
| H3.3 K27M-mutant (+loss of K27me3) | Mutant | |
| H3.3 G34R/V-mutant | Mutant |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurimoto, M.; Rockenbach, Y.; Kato, A.; Natsume, A. Prediction of Tumor Development and Urine-Based Liquid Biopsy for Molecule-Targeted Therapy of Gliomas. Genes 2023, 14, 1201. https://doi.org/10.3390/genes14061201
Kurimoto M, Rockenbach Y, Kato A, Natsume A. Prediction of Tumor Development and Urine-Based Liquid Biopsy for Molecule-Targeted Therapy of Gliomas. Genes. 2023; 14(6):1201. https://doi.org/10.3390/genes14061201
Chicago/Turabian StyleKurimoto, Michihiro, Yumi Rockenbach, Akira Kato, and Atsushi Natsume. 2023. "Prediction of Tumor Development and Urine-Based Liquid Biopsy for Molecule-Targeted Therapy of Gliomas" Genes 14, no. 6: 1201. https://doi.org/10.3390/genes14061201
APA StyleKurimoto, M., Rockenbach, Y., Kato, A., & Natsume, A. (2023). Prediction of Tumor Development and Urine-Based Liquid Biopsy for Molecule-Targeted Therapy of Gliomas. Genes, 14(6), 1201. https://doi.org/10.3390/genes14061201