


Complete Chloroplast Genome Determination of Ranunculus sceleratus from Republic of Korea (Ranunculaceae) and Comparative Chloroplast Genomes of the Members of the Ranunculus Genus



Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Sampling and Sequencing
2.2. Chloroplast Genome Assembly and Annotation
2.3. Simple Sequence Repeat Analysis
2.4. Phylogenetic Analysis
2.5. Comparative CP Genome Analysis and Screening of Barcoding Regions
2.6. Specific Barcode Region Primer Design
2.7. Positive Selection Analysis
3. Results and Discussion
3.1. CP Genome Characterization for Ranunculus Species
3.2. SSR Analysis of Ranunculus CP Genomes
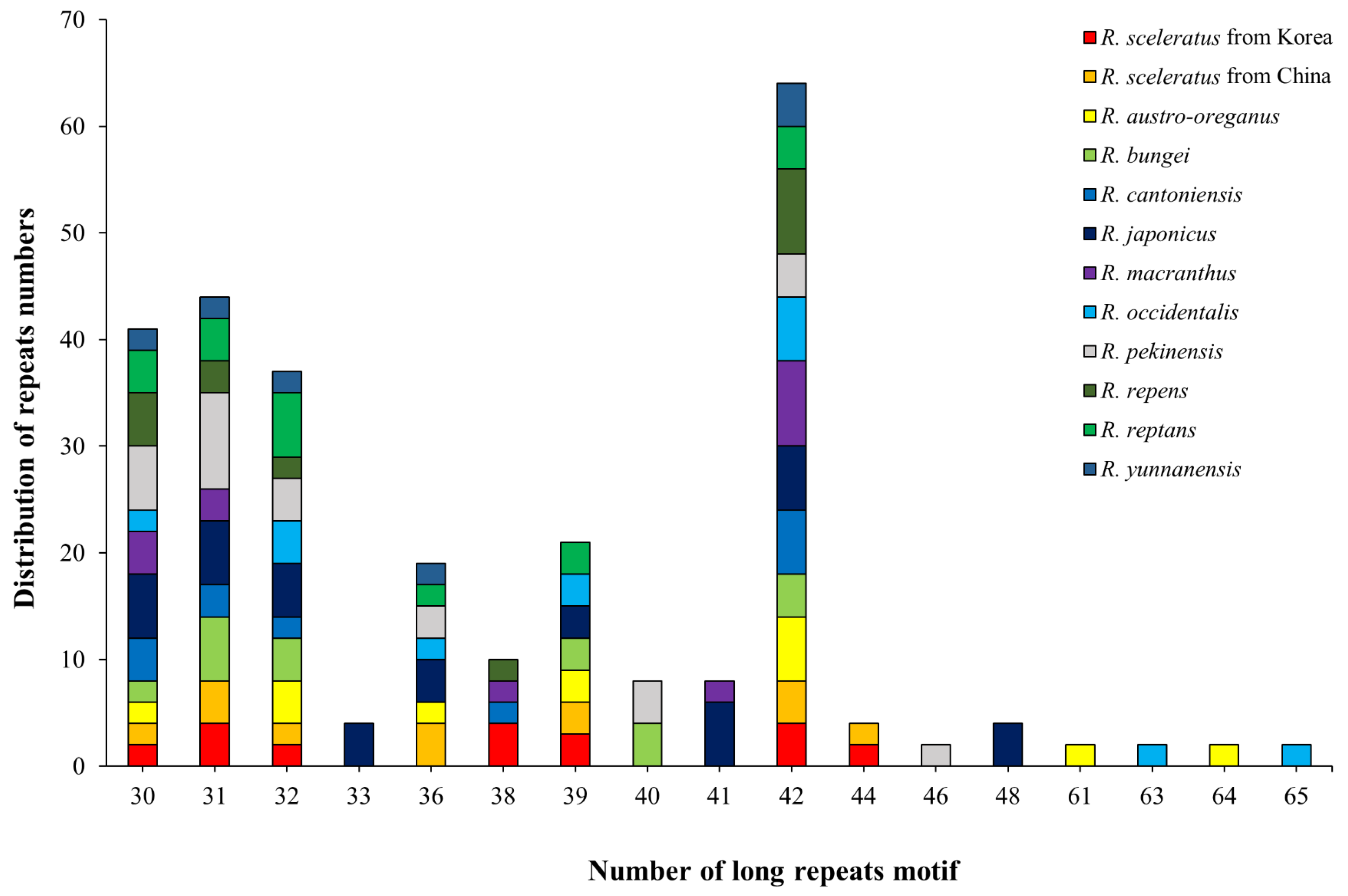
3.2.1. Long Repeats
3.2.2. SSR Analysis
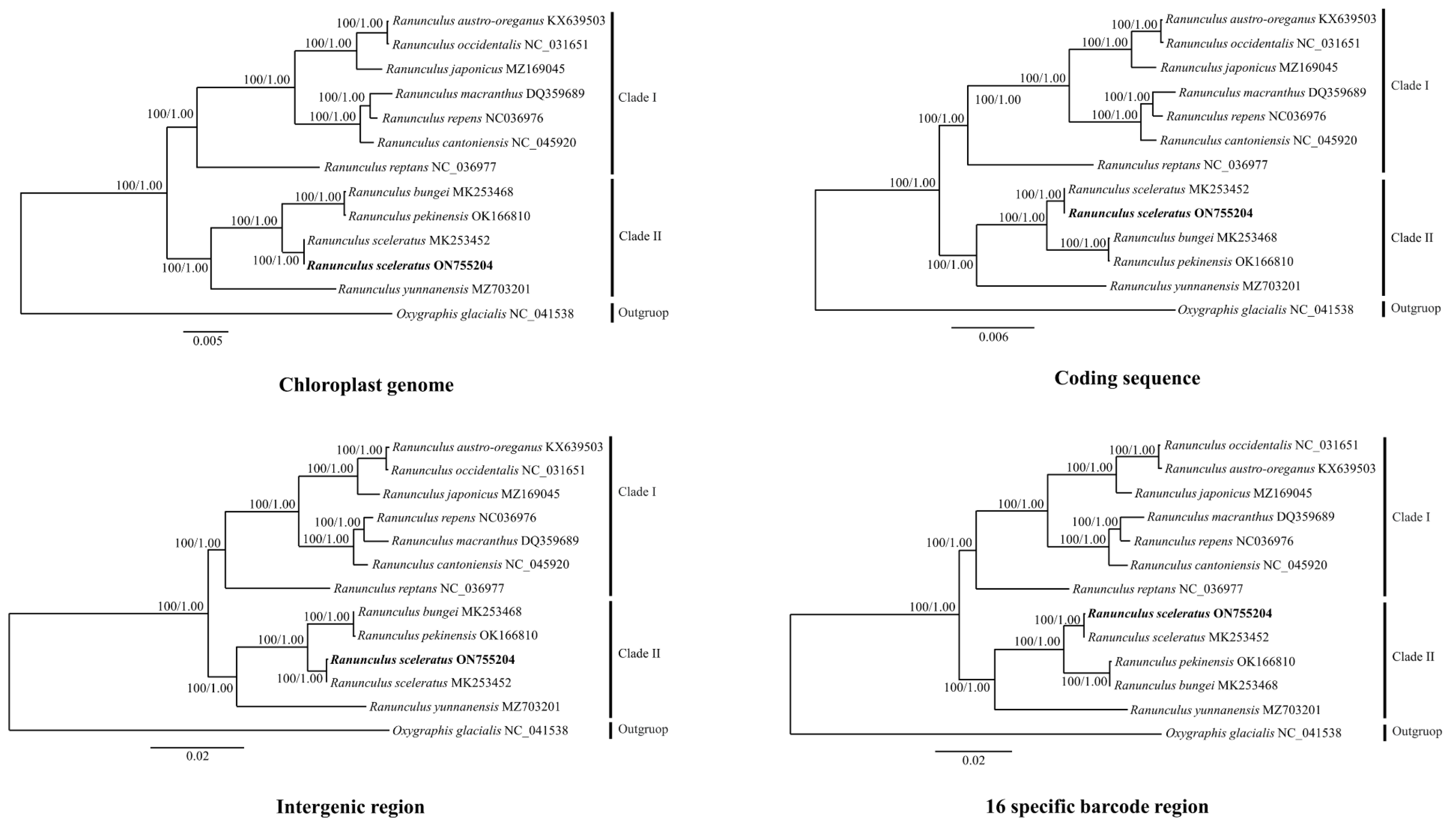
3.3. Phylogenetic Analysis
3.4. Barcoding Region Screening of Genome Divergence Regions
3.5. Barcode Validation of Hotspot Regions in the Genome
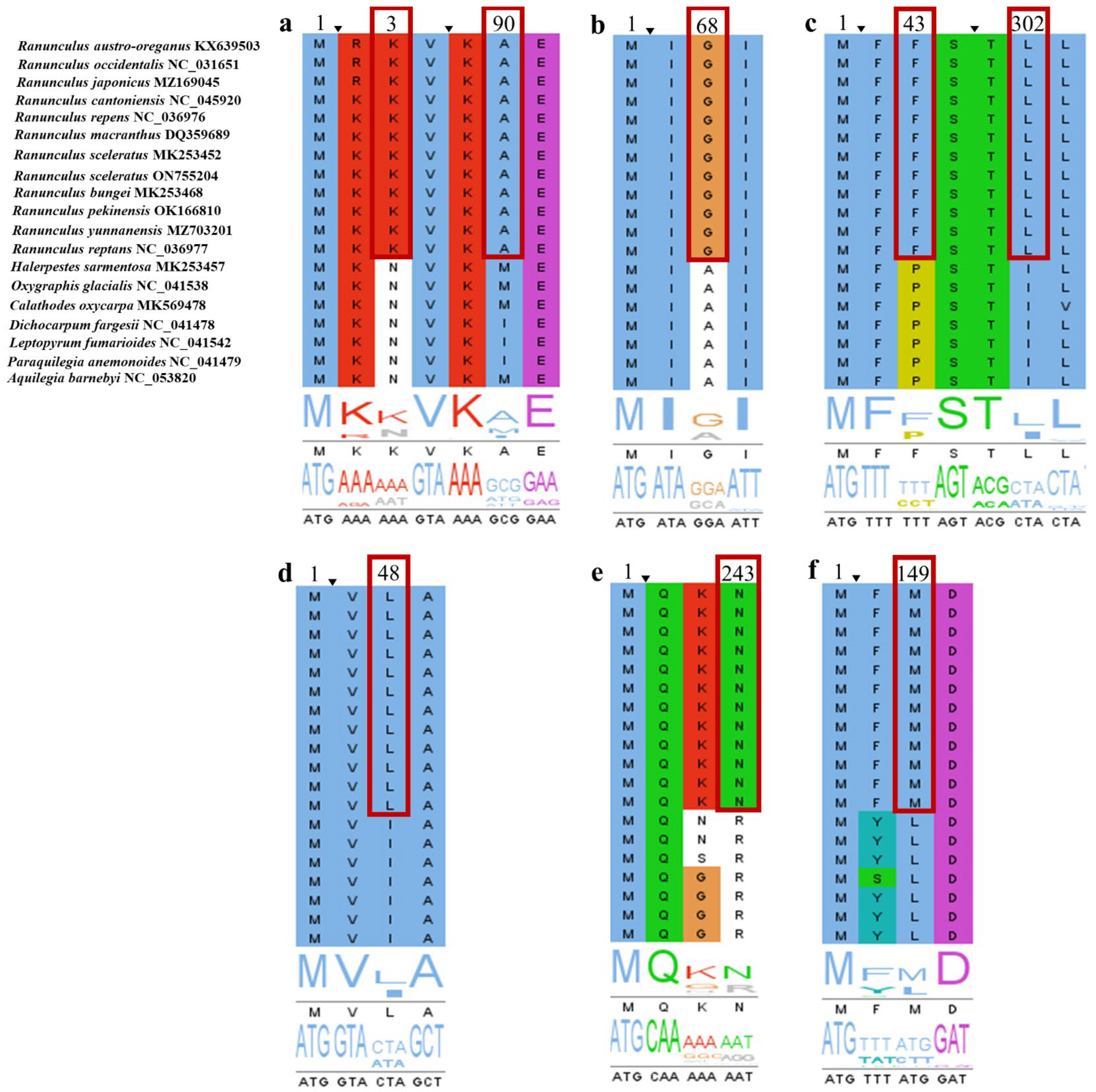
3.6. Positive Selection Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CP | chloroplast genome |
| CDS | coding sequence |
| BEB | Bayes Empirical Bayes |
| NEB | Naïve Empirical Bayes |
| ML | maximum likelihood |
| Pi | nucleotide diversity |
| SSR | simple sequence repeat |
References
- Almerekova, S.; Shchegoleva, N.; Abugalieva, S.; Turuspekov, Y. The molecular taxonomy of three endemic Central Asian species of Ranunculus (Ranunculaceae). PLoS ONE 2020, 15, e0240121. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M. Ranunculaceae. In The Families and Genera of Vascular Plants. II. Dicotyledons; Kubitzki, K., Rower, J.G., Bittrich, V., Eds.; Springer: New York, NY, USA, 1993; pp. 563–583. [Google Scholar]
- Tamura, M. Angiospermae. Ordnung Ranunculales. Fam. Ranunculaceae. II. Systematic Part. In Die Natürliche Pflanzenfamilien, 2nd ed.; Hiepko, P., Ed.; Duncker & Humblot: Berlin, Germany, 1995; pp. 223–519. [Google Scholar]
- Emadzade, K.; Lehnebach, C.; Lockhart, P.; Hörandl, E. A molecular phylogeny, morphology and classification of genera of Ranunculeae (Ranunculaceae). Taxon 2010, 59, 809–828. [Google Scholar] [CrossRef]
- Hoerandl, E.; Emadzade, K. Evolutionary classification: A case study on the diverse plant genus Ranunculus L. (Ranunculaceae). Perspect. Plant Ecol. Evol. Syst. 2012, 14, 310–324. [Google Scholar] [CrossRef]
- Telford, A.; O’Hare, M.T.; Cavers, S.; Holmes, N. Can genetic bar-coding be used to identify aquatic Ranunculus L. subgenus Batrachium (DC) A. Gray? A test using some species from the British Isles. Aquat. Bot. 2011, 95, 65–70. [Google Scholar] [CrossRef]
- Mei, H.; Zuo, S.; Ye, L.; Wang, J.; Ma, S. Review of the application of the traditional Chinese medicinal herb, Ranunculus sceleratus Linn. J. Med. Plant Res. 2012, 6, 1821–1826. [Google Scholar] [CrossRef]
- Zhang, Z.; Miao, Y.; Xu, M.; Cheng, W.; Yang, C.; She, X.; Geng, Q.; Zhang, Q. TianJiu therapy for α-naphthyl isothiocyanate-induced intrahepatic cholestasis in rats treated with fresh Ranunculus sceleratus L. J. Ethnopharmacol. 2020, 248, 112310. [Google Scholar] [CrossRef]
- Li, X.F. The buttercup medical plant resource of Qian Xi Na Zhou. Chin. Wild Plant Resour. 1999, 18, 49–50. [Google Scholar]
- Shahid, S.; Riaz, T.; Asghar, M.N. Screening of Ranunculus sceleratus for enzyme inhibition, antibacterial and antioxidant activities. Bangladesh J. Pharmacol. 2015, 10, 436–442. [Google Scholar] [CrossRef]
- Akkol, E.K.; Süntar, I.; Erdoğan, T.F.; Keleş, H.; Gonenc, T.M.; Kıvçak, B. Wound healing and anti-inflammatory properties of Ranunculus pedatus and Ranunculus constantinapolitanus: A comparative study. J. Ethnopharmacol. 2012, 139, 478–484. [Google Scholar] [CrossRef]
- Cao, B.J.; Meng, Q.Y.; Ji, N. Analgesic and anti-inflammatory effects of Ranunculus japonicus extract. Planta Med. 1992, 58, 496–498. [Google Scholar] [CrossRef]
- Kong, C.S.; Lee, J.I.; Karadeniz, F.; Kim, H.; Seo, Y. Effect of the Arctic terrestrial plant Ranunculus hyperboreus on LPS-induced inflammatory response via MAPK pathways. Z. Naturforsch. C 2018, 73, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhou, C.X.; Pan, Y.; Gao, X.; Wu, X.; Bai, H.; Zhou, L.; Chen, Z.; Zhang, S.; Shi, S.; et al. Evaluation of antiviral activity of compounds isolated from Ranunculus sieboldii and Ranunculus sceleratus. Planta Med. 2005, 71, 1128–1133. [Google Scholar] [CrossRef] [PubMed]
- Saber, A.H.; Mahran, G.H.; El–Alfy, T. Phytochemical investigation of Ranunculus sceleratus L. Planta Med. 1968, 16, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.S.; Dinzouna-Boutamba, S.D.; Lee, S.; Moon, Z.; Kwak, D.; Rhee, M.H.; Chung, D.I.; Hong, Y.; Goo, Y.K. Antimalarial effect of the total glycosides of the medicinal plant, Ranunculus japonicus. Pathogens 2021, 10, 532. [Google Scholar] [CrossRef]
- Goo, Y.K. Therapeutic potential of Ranunculus species (Ranunculaceae): A literature review on traditional medicinal herbs. Plants 2022, 11, 1599. [Google Scholar] [CrossRef]
- Dong, T.T.; Cui, X.M.; Song, Z.H.; Zhao, K.J.; Ji, Z.N.; Lo, C.K.; Tsim, K.W. Chemical assessment of roots of Panax notoginseng in China: Regional and seasonal variations in its active constituents. J. Agric. Food Chem. 2003, 51, 4617–4623. [Google Scholar] [CrossRef]
- Lian, C.; Yang, H.; Lan, J.; Zhang, X.; Zhang, F.; Yang, J.; Chen, S. Comparative analysis of chloroplast genomes reveals phylogenetic relationships and intraspecific variation in the medicinal plant Isodon rubescens. PLoS ONE 2022, 17, e0266546. [Google Scholar] [CrossRef]
- Guo, B.J. A technique for monitoring pollutants by means of micronuclear cells in root-tip cells of Ranunculus sceleratus. Acta Sci. Circum. 1985, 5, 322–326. [Google Scholar]
- Hu, J.F.; Wang, X.M.; Shi, G.R.; Hu, Z.D. The purification of Fe2+ in wastewater with Ranunculus sceleratus in gardens. J. Huaibei Coal Indust. Teach. College 2007, 28, 31–33. [Google Scholar]
- Wang, D.; Zhang, Y.L.; Pang, B. Study on purification effect in different concentrations of sewage by Ranunculus sceleratus L. J. Shandong For. Sci. Technol. 2009, 39, 14–16. [Google Scholar]
- Fazekas, A.J.; Burgess, K.S.; Kesanakurti, P.R.; Graham, S.W.; Newmaster, S.G.; Husband, B.C.; Percy, D.M.; Hajibabaei, M.; Barrett, S.C. Multiple multilocus DNA barcodes from the plastid genome discriminate plant species equally well. PLoS ONE 2008, 3, e2802. [Google Scholar] [CrossRef] [PubMed]
- Kress, W.J.; Wurdack, K.J.; Zimmer, E.A.; Weigt, L.A.; Janzen, D.H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. USA 2005, 102, 8369–8374. [Google Scholar] [CrossRef]
- Hollingsworth, M.L.; Clark, A.A.; Forrest, L.L.; Richardson, J.; Pennington, R.T.; Long, D.G.; Cowan, R.; Chase, M.W.; Gaudeul, M.; Hollingsworth, P.M. Selecting barcoding loci for plants: Evaluation of seven candidate loci with species-level sampling in three divergent groups of land plants. Mol. Ecol. Res. 2009, 9, 439–457. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Zeng, Z.-F.; Du, Y.; Mao, C.-M.; Xie, L.; Guo, X.-L.; Yue, L.L. The complete chloroplast genome of Ranunculus yunnanensis (Ranunculaceae). Mitochondrial DNA Part B Resour. 2022, 7, 60–61. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Li, L.; Quan, H.; Yang, J.; Zhang, Z.; Liao, Z.; Lan, X. Comparative analyses of chloroplast genomes from 14 Zanthoxylum species: Identification of variable DNA markers and phylogenetic relationships within the genus. Front. Plant Sci. 2021, 11, 605793. [Google Scholar] [CrossRef] [PubMed]
- Lahaye, R.; Van der Bank, M.; Bogarin, D.; Warner, J.; Pupulin, F.; Gigot, G.; Maurin, O.; Duthoit, S.; Barraclough, T.G.; Savolainen, V. DNA barcoding the floras of biodiversity hotspots. Proc. Natl. Acad. Sci. USA 2008, 105, 2923–2928. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Henry, R.J.; Rossetto, M.; Wang, Y.; Chen, S. Plant DNA barcoding: From gene to genome. Biol. Rev. 2015, 90, 157–166. [Google Scholar] [CrossRef]
- Chase, M.W.; Cowan, R.S.; Hollingsworth, P.M.; van den Berg, C.; Madrinan, S.; Petersen, G.; Seberg, O.; Jørgsensen, T.; Cameron, K.M.; Carine, M.; et al. A proposal for a standardised protocol to barcode all land plants. Taxon 2007, 56, 295–299. [Google Scholar] [CrossRef]
- Li, T.; Fu, X.; Deng, H.; Han, X.; Wen, F.; Xu, L. The complete chloroplast genome of Ranunculus cantoniensis. Mitochondrial DNA Part B Resour. 2019, 4, 1095–1096. [Google Scholar] [CrossRef]
- Liu, X.; Yang, W.; Zhang, D.; Liu, H.; Cui, Y.; Wang, L.; Hu, H.; Yin, Y.; Zhang, G. The complete chloroplast genome sequence of Ranunculus pekinensis (L. Liou) Luferov (Ranunculaceae), a species endemic to China. Mitochondrial DNA Part B Resour. 2022, 7, 841–843. [Google Scholar] [CrossRef]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.-Q.; Yan, H.-J.; Wu, Y.-Z.; Wang, R.; Xiao, X.-F.; Qi, Z.-C.; Yan, X.L. The complete chloroplast genome sequence of Japanese buttercup Ranunculus japonicus Thunb. Mitochondrial DNA Part B Resour. 2021, 6, 3186–3187. [Google Scholar] [CrossRef] [PubMed]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; DePamphilis, C.W.; Yi, T.-S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Sys. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 1997, 13, 555–556. [Google Scholar] [CrossRef]
- Yang, Z.; Dos Reis, M. Statistical properties of the branch-site test of positive selection. Mol. Biol. Evol. 2010, 28, 1217–1228. [Google Scholar] [CrossRef]
- Lan, Y.; Sun, J.; Tian, R.; Bartlett, D.H.; Li, R.; Wong, Y.H.; Zhang, W.; Qiu, J.W.; Xu, T.; He, L.S.; et al. Molecular adaptation in the world’s deepest-living animal: Insights from transcriptome sequencing of the hadal amphipod Hirondellea gigas. Mol. Ecol. 2017, 26, 3732–3743. [Google Scholar] [CrossRef]
- Xie, D.-F.; Yu, Y.; Deng, Y.-Q.; Li, J.; Liu, H.-Y.; Zhou, S.-D.; He, X.J. Comparative analysis of the chloroplast genomes of the Chinese endemic genus Urophysa and their contribution to chloroplast phylogeny and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 1847. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 2002, 19, 908–917. [Google Scholar] [CrossRef]
- Clamp, M.; Cuff, J.; Searle, S.M.; Barton, G.J. The jalview java alignment editor. Bioinformatics 2004, 20, 426–427. [Google Scholar] [CrossRef]
- Dann, M.; Bellot, S.; Schepella, S.; Schaefer, H.; Tellier, A. Mutation rates in seeds and seed-banking influence substitution rates across the angiosperm phylogeny. bioRxiv 2017. [Google Scholar] [CrossRef]
- He, J.; Yao, M.; Lyu, R.-D.; Lin, L.-L.; Liu, H.-J.; Pei, L.-Y.; Yan, S.X.; Xie, L.; Cheng, J. Structural variation of the complete chloroplast genome and plastid phylogenomics of the genus Asteropyrum (Ranunculaceae). Sci. Rep. 2019, 9, 15285. [Google Scholar] [CrossRef] [PubMed]
- Hirao, T.; Watanabe, A.; Kurita, M.; Kondo, T.; Takata, K. Complete nucleotide sequence of the Cryptomeria japonica D. Don. chloroplast genome and comparative chloroplast genomics: Diversified genomic structure of coniferous species. BMC Plant Biol. 2008, 8, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Zhou, T.; Han, K.; Yang, Y.; Zhao, J.; Liu, Z.L. The complete chloroplast genome sequences of six Rehmannia species. Genes 2017, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Dobrogojski, J.; Adamiec, M.; Luciński, R. The chloroplast genome: A review. Acta Physiol. Plant. 2020, 42, 1–13. [Google Scholar] [CrossRef]
- Ravi, V.; Khurana, J.P.; Tyagi, A.K.; Khurana, P. An update on chloroplast genomes. Plant Syst. Evol. 2008, 271, 101–122. [Google Scholar] [CrossRef]
- Sammut, R.; Huttley, G. Regional context in the alignment of biological sequence pairs. J. Mol. Evol. 2011, 72, 147–159. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, J.G.; Chen, X.L.; Cui, Y.X.; Xu, Z.C.; Li, Y.H.; Song, J.Y.; Duan, B.Z.; Yao, H. Gene losses and partial deletion of small single-copy regions of the chloroplast genomes of two hemiparasitic Taxillus species. Sci. Rep. 2017, 7, 12834. [Google Scholar] [CrossRef]
- Magee, A.M.; Aspinall, S.; Rice, D.W.; Cusack, B.P.; Sémon, M.; Perry, A.S.; Stefanović, S.; Milbourne, D.; Barth, S.; Palmer, J.D.; et al. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res. 2010, 20, 1700–1710. [Google Scholar] [CrossRef]
- McCoy, S.R.; Kuehl, J.V.; Boore, J.L.; Raubeson, L.A. The complete plastid genome sequence of Welwitschia mirabilis: An unusually compact plastome with accelerated divergence rates. BMC Evol. Biol. 2008, 8, 1–16. [Google Scholar] [CrossRef]
- Gao, L.; Yi, X.; Yang, Y.-X.; Su, Y.-J.; Wang, T. Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: Insights into evolutionary changes in fern chloroplast genomes. BMC Evol. Biol. 2009, 9, 1–14. [Google Scholar] [CrossRef]
- Guisinger, M.M.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: Rearrangements, repeats, and codon usage. Mol. Biol. Evol. 2011, 28, 583–600. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, J.; Luo, L.; Wei, X.; Zhang, J.; Qi, Y.; Zhang, B.; Liu, H.; Xiao, P. Complete chloroplast genome sequences of Schisandra chinensis: Genome structure, comparative analysis, and phylogenetic relationship of basal angiosperms. Sci. China Life Sci. 2017, 60, 1286–1290. [Google Scholar] [CrossRef] [PubMed]
- Wen, F.; Wu, X.; Li, T.; Jia, M.; Liu, X.; Liao, L. The complete chloroplast genome of Stauntonia chinensis and compared analysis revealed adaptive evolution of subfamily Lardizabaloideae species in China. BMC Genom. 2021, 22, 161. [Google Scholar] [CrossRef] [PubMed]
- Ambreen, H.; Kumar, S.; Variath, M.T.; Joshi, G.; Bali, S.; Agarwal, M.; Kumar, A.; Jagannath, A.; Goel, S. Development of genomic microsatellite markers in Carthamus tinctorius L.(safflower) using next generation sequencing and assessment of their cross-species transferability and utility for diversity analysis. PLoS ONE 2015, 10, e0135443. [Google Scholar] [CrossRef] [PubMed]
- Martina, M.; Acquadro, A.; Barchi, L.; Gulino, D.; Brusco, F.; Rabaglio, M.; Portis, F.; Portis, E.; Lanteri, S. Genome-wide survey and development of the first microsatellite markers database (AnCorDB) in Anemone coronaria L. Int. J. Mol. Sci. 2022, 23, 3126. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Peng, Z.; Sun, H.; Yue, X.; Lou, Y.; Dong, L.; Wang, L.; Gao, Z. Developing genome-wide microsatellite markers of bamboo and their applications on molecular marker assisted taxonomy for accessions in the genus Phyllostachys. Sci. Rep. 2015, 5, 8018. [Google Scholar] [CrossRef]
- Alzahrani, D.A.; Albokhari, E.J.; Yaradua, S.S.; Abba, A. Comparative analysis of chloroplast genomes of four medicinal Capparaceae species: Genome structures, phylogenetic relationships and adaptive evolution. Plants 2021, 10, 1229. [Google Scholar] [CrossRef]
- Song, Y.; Wang, S.; Ding, Y.; Xu, J.; Li, M.F.; Zhu, S.; Chen, N. Chloroplast genomic resource of Paris for species discrimination. Sci. Rep. 2017, 7, 3427. [Google Scholar] [CrossRef]
- Wiegleb, G.; Bobrov, A.A.; Zalewska-Gałosz, J. A taxonomic account of Ranunculus section Batrachium (Ranunculaceae). Phytotaxa 2017, 319, 1–55. [Google Scholar] [CrossRef]
- Liu, H.; He, J.; Ding, C.; Lyu, R.; Pei, L.; Cheng, J.; Xie, L. Comparative analysis of complete chloroplast genomes of Anemoclema, Anemone, Pulsatilla, and Hepatica revealing structural variations among genera in tribe Anemoneae (Ranunculaceae). Front. Plant Sci. 2018, 9, 1097. [Google Scholar] [CrossRef]
- Yu, X.-Q.; Drew, B.T.; Yang, J.-B.; Gao, L.-M.; Li, D.-Z. Comparative chloroplast genomes of eleven Schima (Theaceae) species: Insights into DNA barcoding and phylogeny. PLoS ONE 2017, 12, e0178026. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xing, Y.; Xu, L.; Bao, G.; Zhan, Z.; Yang, Y.; Wang, J.; Li, S.; Zhang, D.; Kang, T. Comparative analysis of the complete chloroplast genome sequences of six species of Pulsatilla Miller, Ranunculaceae. Chin. Med. 2019, 14, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Doorduin, L.; Gravendeel, B.; Lammers, Y.; Ariyurek, Y.; Chin-A-Woeng, T.; Vrieling, K. The complete chloroplast genome of 17 individuals of pest species Jacobaea vulgaris: SNPs, microsatellites and barcoding markers for population and phylogenetic studies. DNA Res. 2011, 18, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.; Fahmy, R.M.; Mosa, K.A.; Helmy, M.; El-Feky, F.A. Identification of effective DNA barcodes for Triticum plants through chloroplast genome-wide analysis. Comput. Biol. Chem. 2017, 71, 20–31. [Google Scholar] [CrossRef]
- Wang, W.; Chen, S.; Zhang, X. Whole-genome comparison reveals divergent IR borders and mutation hotspots in chloroplast genomes of herbaceous bamboos (Bambusoideae: Olyreae). Molecules 2018, 23, 1537. [Google Scholar] [CrossRef]
- Yan, H.-F.; Liu, Y.-J.; Xie, X.-F.; Zhang, C.-Y.; Hu, C.-M.; Hao, G.; Ge, X.J. DNA barcoding evaluation and its taxonomic implications in the species-rich genus Primula L. in China. PLoS ONE 2015, 10, e0122903. [Google Scholar] [CrossRef]
- Hudson, G.S.; Mason, J.G. The chloroplast genes encoding subunits of the H+-ATP synthase. Photosynth. Res. 1988, 18, 205–222. [Google Scholar] [CrossRef]
- Hajdukiewicz, P.T.; Allison, L.A.; Maliga, P. The two RNA polymerases encoded by the nuclear and the plastid compartments transcribe distinct groups of genes in tobacco plastids. EMBO J. 1997, 16, 4041–4048. [Google Scholar] [CrossRef]
- Piot, A.; Hackel, J.; Christin, P.A.; Besnard, G. One-third of the plastid genes evolved under positive selection in PACMAD grasses. Planta 2018, 247, 255–266. [Google Scholar] [CrossRef]
- Yu, Q.-B.; Huang, C.; Yang, Z.-N. Nuclear-encoded factors associated with the chloroplast transcription machinery of higher plants. Front. Plant Sci. 2014, 5, 316. [Google Scholar] [CrossRef]
- Fromont-Racine, M.; Senger, B.; Saveanu, C.; Fasiolo, F. Ribosome assembly in eukaryotes. Gene 2003, 313, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Moin, M.; Bakshi, A.; Madhav, M.S.; Kirti, P.B. Expression profiling of ribosomal protein gene family in dehydration stress responses and characterization of transgenic rice plants overexpressing RPL23A for water-use efficiency and tolerance to drought and salt stresses. Front. Chem. 2017, 5, 97. [Google Scholar] [CrossRef] [PubMed]
- Casano, L.M.; Zapata, J.M.; Martı́n, M.; Sabater, B. Chlororespiration and poising of cyclic electron transport: Plastoquinone as electron transporter between thylakoid NADH dehydrogenase and peroxidase. J. Biol. Chem. 2000, 275, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Kofer, W.; Koop, H.-U.; Wanner, G.; Steinmüller, K. Mutagenesis of the genes encoding subunits A, C, H, I, J and K of the plastid NAD (P) H-plastoquinone-oxidoreductase in tobacco by polyethylene glycol-mediated plastome transformation. Mol. Gen. Genet. 1998, 258, 166–173. [Google Scholar] [CrossRef]
- Sabater, B. On the edge of dispensability, the chloroplast ndh genes. Int. J. Mol. Sci. 2021, 22, 12505. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.; Sabater, B. Plastid ndh genes in plant evolution. Plant Physiol. Biochem. 2010, 48, 636–645. [Google Scholar] [CrossRef]
- Kulheim, C.; Ågren, J.; Jansson, S. Rapid regulation of light harvesting and plant fitness in the field. Science 2002, 297, 91–93. [Google Scholar] [CrossRef]
- Martín, M.; Funk, H.T.; Serrot, P.H.; Poltnigg, P.; Sabater, B. Functional characterization of the thylakoid Ndh complex phosphorylation by site-directed mutations in the ndhF gene. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2009, 1787, 920–928. [Google Scholar] [CrossRef]
- Terashima, I.; Araya, T.; Miyazawa, S.-I.; Sone, K.; Yano, S. Construction and maintenance of the optimal photosynthetic systems of the leaf, herbaceous plant and tree: An eco-developmental treatise. Ann. Bot. 2005, 95, 507–519. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Reads | 958,608,148 |
| Percentage of subsampling reads (%) | 15.0 |
| GC content of total reads (%) | 41.22 |
| Chloroplast genome length (bp) | 156,329 |
| Large single-copy length (bp) | 85,840 |
| Small single-copy length (bp) | 19,885 |
| Inverted repeat length (bp) | 25,302 |
| Total number of genes | 112 |
| No. | Region Name | Pi | Alignment Length (bp) | ML Tree (%) | BLAST (%) | Total Identification Rate (%) |
|---|---|---|---|---|---|---|
| 1 | petG-trnW-CCA | 0.091 | 175 | 75 | 83.3 | 79.2 |
| 2 | rpl32-trnL-UAG | 0.083 | 885 | 83.3 | 83.3 | 83.3 |
| 3 | rpl16-rps3 | 0.073 | 217 | 83.3 | 83.3 | 83.3 |
| 4 | rps8-rpl14 | 0.069 | 335 | 91.7 | 91.7 | 91.7 |
| 5 | rps16-trnQ-UUG | 0.067 | 1237 | 83.3 | 83.3 | 83.3 |
| 6 | ndhG-ndhI | 0.062 | 409 | 83.3 | 100 | 91.7 |
| 7 | petN-psbM | 0.059 | 1327 | 91.7 | 100 | 95.9 |
| 8 | accD-psaI | 0.055 | 819 | 83.3 | 83.3 | 83.3 |
| 9 | trnG-GCC-trnfM-CAU | 0.054 | 209 | 83.3 | 83.3 | 83.3 |
| 10 | atpF-atpI | 0.053 | 1956 | 91.7 | 100 | 95.9 |
| 11 | trnT-UGU-trnL-UAA | 0.052 | 995 | 83.3 | 83.3 | 83.3 |
| 12 | psbZ-trnG-GCC | 0.052 | 424 | 83.3 | 83.3 | 83.3 |
| 13 | trnK-UUU-rps16 | 0.051 | 565 | 83.3 | 83.3 | 83.3 |
| 14 | ndhC-trnV-UAC | 0.049 | 1441 | 100 | 100 | 100.0 |
| 15 | trnT-GGU-psbD | 0.041 | 1589 | 91.7 | 100 | 95.9 |
| 16 | psbE-petL | 0.040 | 1274 | 100 | 100 | 100.0 |
| No. | Region Name | Length (bp) | ML Tree (%) | BLAST (%) | Total Identification Rate (%) |
|---|---|---|---|---|---|
| 1 | matK | 1527 | 83 | 100 | 91.5 |
| 2 | rbcL | 1428 | 75 | 75 | 75 |
| 3 | psbA-trnH-GUG | 366 | 75 | 75 | 75 |
| 4 | trnL-UAA-trnF-GAA | 418 | 75 | 75 | 75 |
| No. | Intergenic Region | Forward Primer (5′–3′) | Reverse Primer (5′–3′) | Product Size Range (bp) | Annealing Temperature (°C) | Sequencing Success (%) |
|---|---|---|---|---|---|---|
| 1 | petG-trnW-CCA | TACAGACGCGGTGATCAGTTGGAC | CCAAAACCCGATGTCGTAGGTTC | 200–250 | 58 | 100 |
| 2 | rpl32-trnL-UAG | GCAAAATCTATTTCCACCGGGAAT | GTCTACCGATTTCACCATAGCG | 800–900 | 58 | 100 |
| 3 | rpl16-rps3 | CAACGAGTCACACACTAAGCA | ACTATCTATGGGGCATTAGGAA | 200–300 | 58 | 100 |
| 4 | rps8-rpl14 | TCCATGTCAGCATTTCGTATCG | GTGCAATCGCTCGAGAGTTGA | 370–410 | 58 | 100 |
| 5 | rps16-trnQ-UUG | CGCACGTTGCTTTCTACCACA | CGAATCCTTCCGTCCCAGAG | 1000–1200 | 58 | 100 |
| 6 | ndhG-ndhI | GGTCGGTTACCAATGTCAGTGA | AAGGAGCTGTGCAGCAGCGA | 600–700 | 58 | 100 |
| 7 | petN-psbM | GGCTGCTTTAATGGTAGTCTTTAC | TCGCATTCATTGCTACTGCACTG | 1200–1300 | 58 | 100 |
| 8 | accD-psaI | GAGTGAGTTATTTCAGCTTCACG | GGAGGGTAAGTTGAAAGTTGTCAT | 750–850 | 58 | 100 |
| 9 | trnG-GCC-trnfM-CAU | CGATTCCCGCTATCCGCCTA | GGTAGCTCGCAAGGCTCATAAC | 250–300 | 58 | 100 |
| 10 | atpF-atpI | GGCCAGTGACCCAAGGAAAC | TAGGGGAATCCATGGAGGGTCA | 300–1900 | 58 | 100 |
| 11 | trnT-UGU-trnL-UAA | CGGCTATCGGAATCGAACCG | GCGTCTACCAATTTCGCCATATC | 800–1000 | 58 | 100 |
| 12 | psbZ-trnG-GCC | CCCGTTGTATTTGCTTCTTCTGA | CCGCGTCTTCTCCTTGGCAA | 500–600 | 58 | 100 |
| 13 | trnK-UUU-rps16 | AGCCGCACTTAAAAGCCGAGTA | CGATCCCGAAGAGAGGGAAG | 500–700 | 58 | 100 |
| 14 | ndhC-trnV-UAC | GAGTTTCTCTGGCCCTTCATTA | TACCGAGAAGGTCTACGGTTC | 1300–1400 | 58 | 100 |
| 15 | trnT-GGU-psbD | GGCGTAAGTCATCGGTTCAAAT | TCTCCGTAACCAGTCATCCATA | 1500–1600 | 58 | 100 |
| 16 | psbE-petL | CGTGCTTCCAGACATGCTGA | GCCGTATCTTGCTCAGACCAAT | 1200–1300 | 58 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.-R.; Park, S.Y.; Kim, H.; Hong, J.M.; Kim, S.-Y.; Yu, J.-N. Complete Chloroplast Genome Determination of Ranunculus sceleratus from Republic of Korea (Ranunculaceae) and Comparative Chloroplast Genomes of the Members of the Ranunculus Genus. Genes 2023, 14, 1149. https://doi.org/10.3390/genes14061149
Kim K-R, Park SY, Kim H, Hong JM, Kim S-Y, Yu J-N. Complete Chloroplast Genome Determination of Ranunculus sceleratus from Republic of Korea (Ranunculaceae) and Comparative Chloroplast Genomes of the Members of the Ranunculus Genus. Genes. 2023; 14(6):1149. https://doi.org/10.3390/genes14061149
Chicago/Turabian StyleKim, Kang-Rae, So Young Park, Heesoo Kim, Jeong Min Hong, Sun-Yu Kim, and Jeong-Nam Yu. 2023. "Complete Chloroplast Genome Determination of Ranunculus sceleratus from Republic of Korea (Ranunculaceae) and Comparative Chloroplast Genomes of the Members of the Ranunculus Genus" Genes 14, no. 6: 1149. https://doi.org/10.3390/genes14061149
APA StyleKim, K.-R., Park, S. Y., Kim, H., Hong, J. M., Kim, S.-Y., & Yu, J.-N. (2023). Complete Chloroplast Genome Determination of Ranunculus sceleratus from Republic of Korea (Ranunculaceae) and Comparative Chloroplast Genomes of the Members of the Ranunculus Genus. Genes, 14(6), 1149. https://doi.org/10.3390/genes14061149