
Gene Expression Analysis Reveals Potential Regulatory Factors Response to Temperature Stress in Bemisia tabaci Mediterranean



Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Insects
2.2. RNA Extraction
2.3. Library Construction
2.4. Transcriptome Assembly and Gene Functional Annotation
2.5. Alternative Splicing Analysis
2.6. Differential Gene Expression and Functional Enrichment Analysis
2.7. Bioinformatics Analysis
3. Results
3.1. Sequence Assembly and Functional Annotation
3.2. Alternative Splicing
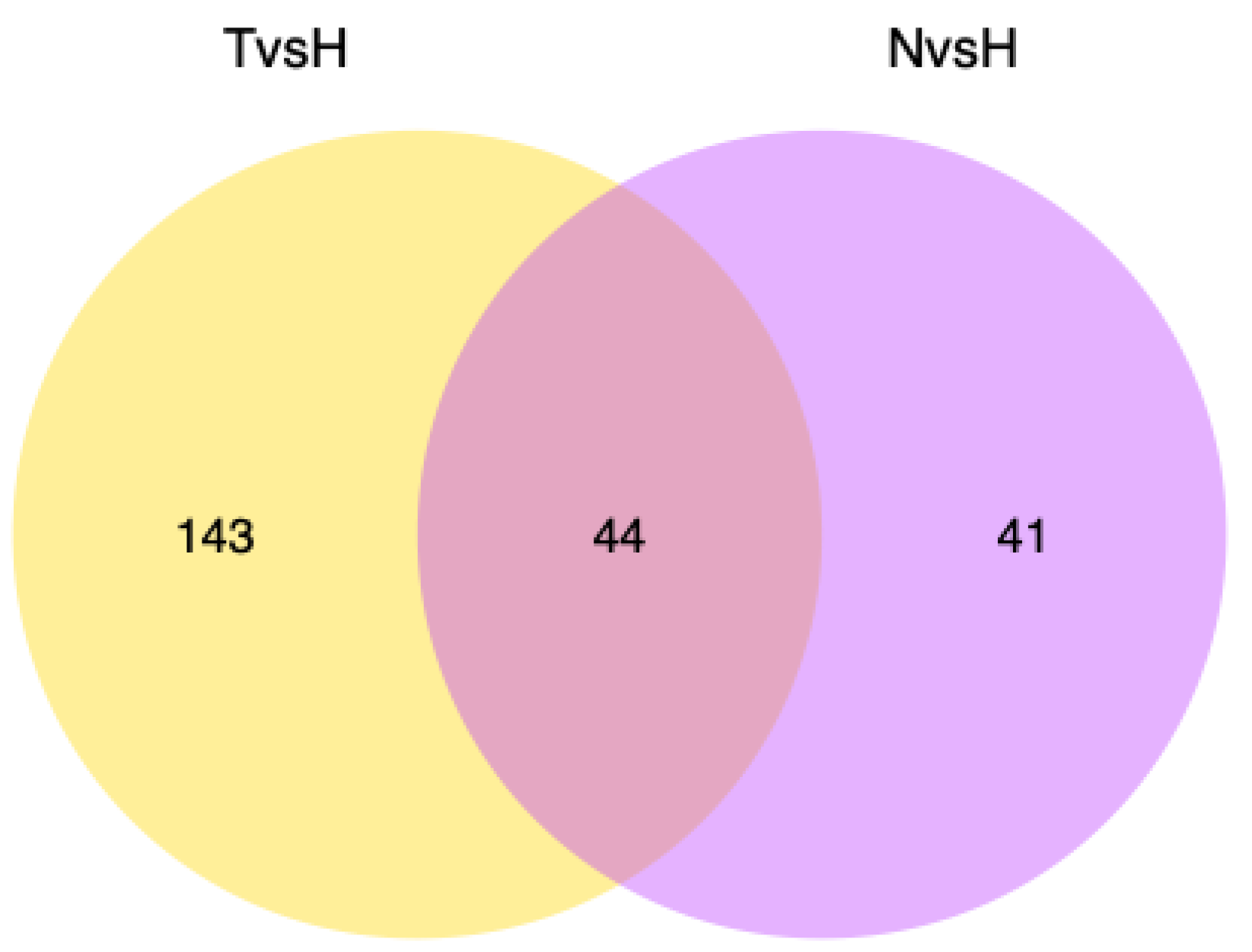
3.3. Expression Differences between High- and Low-Temperature Populations
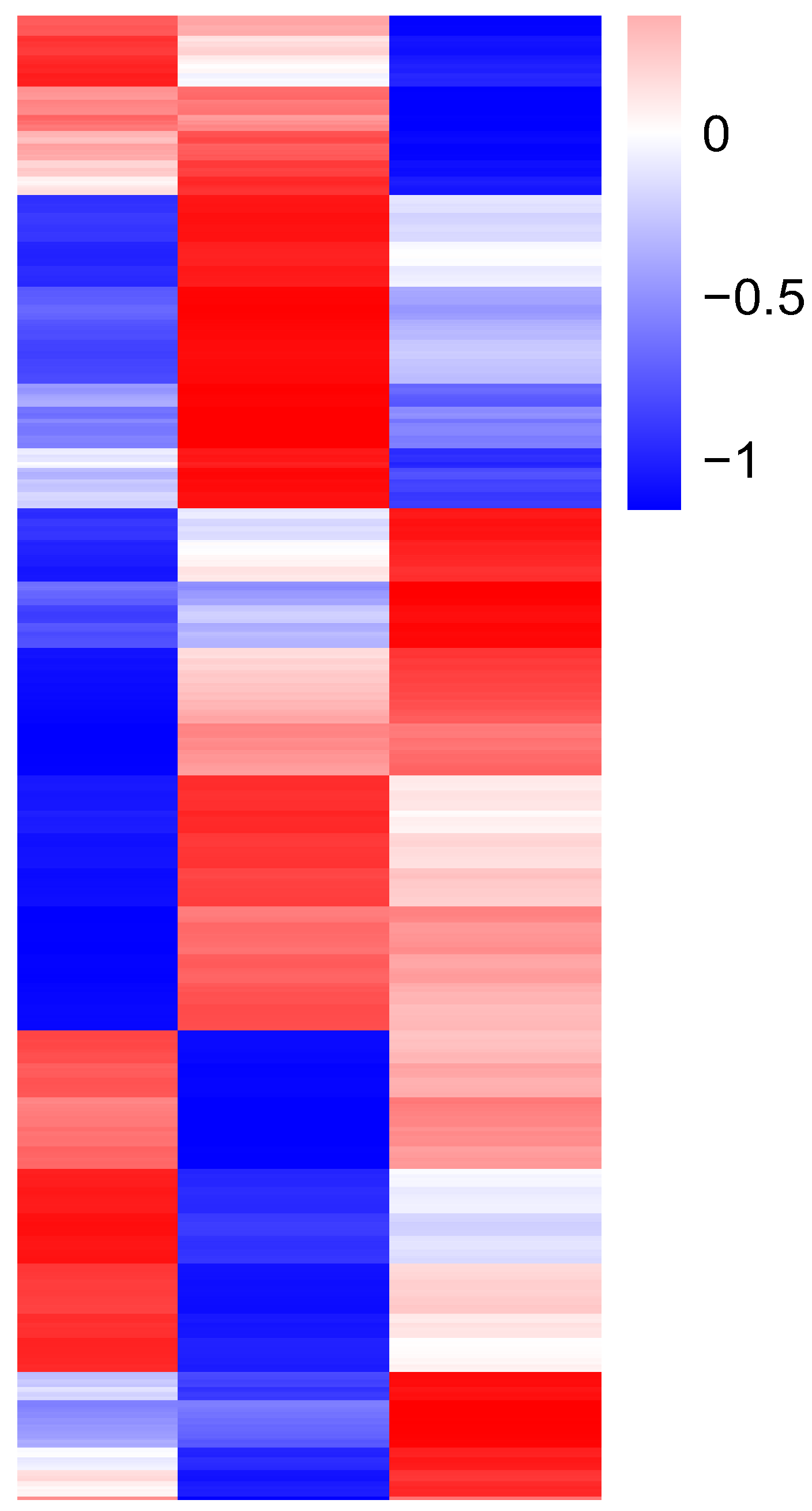
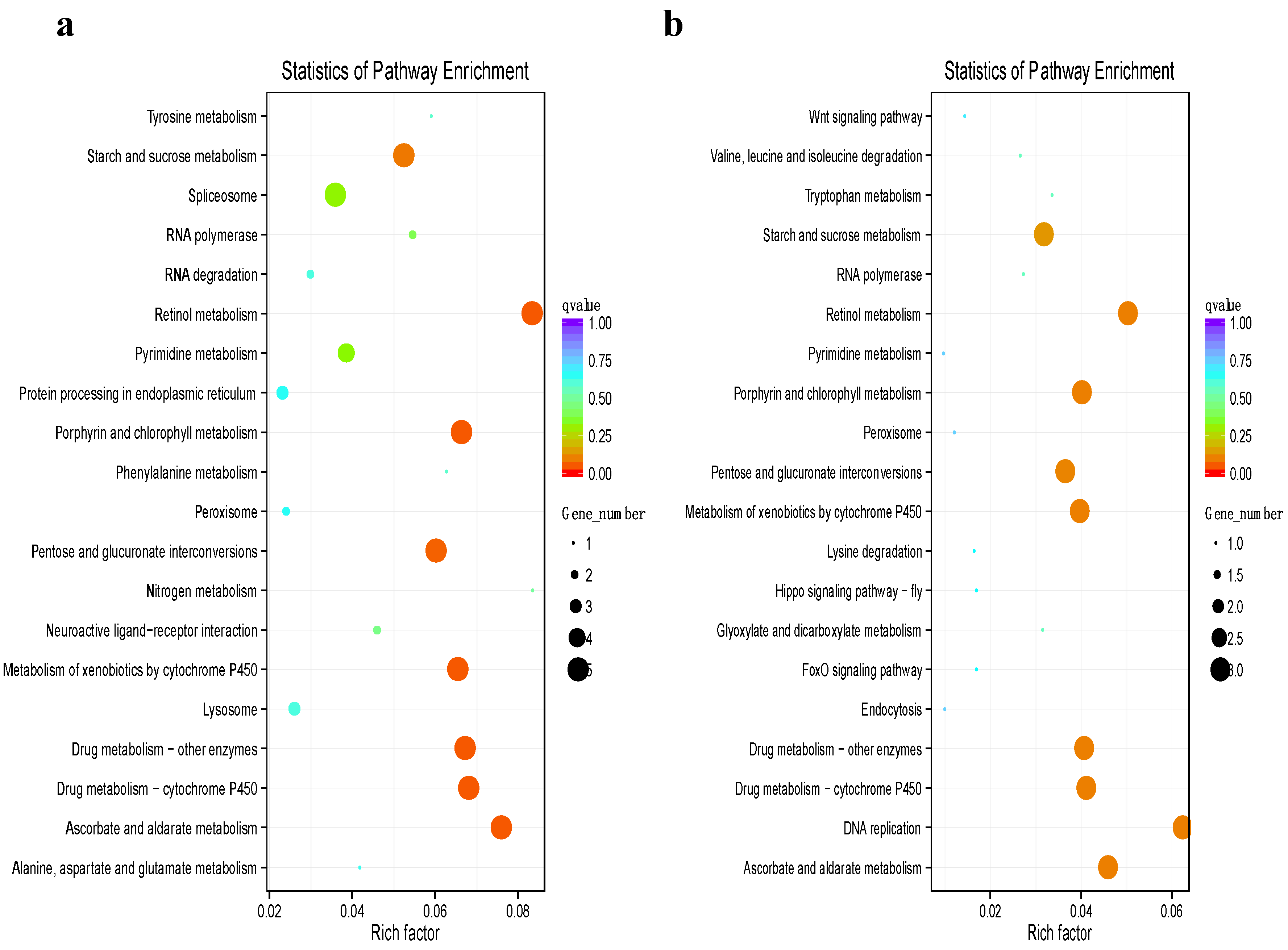
3.4. Functional Annotation of Differentially Expressed Genes
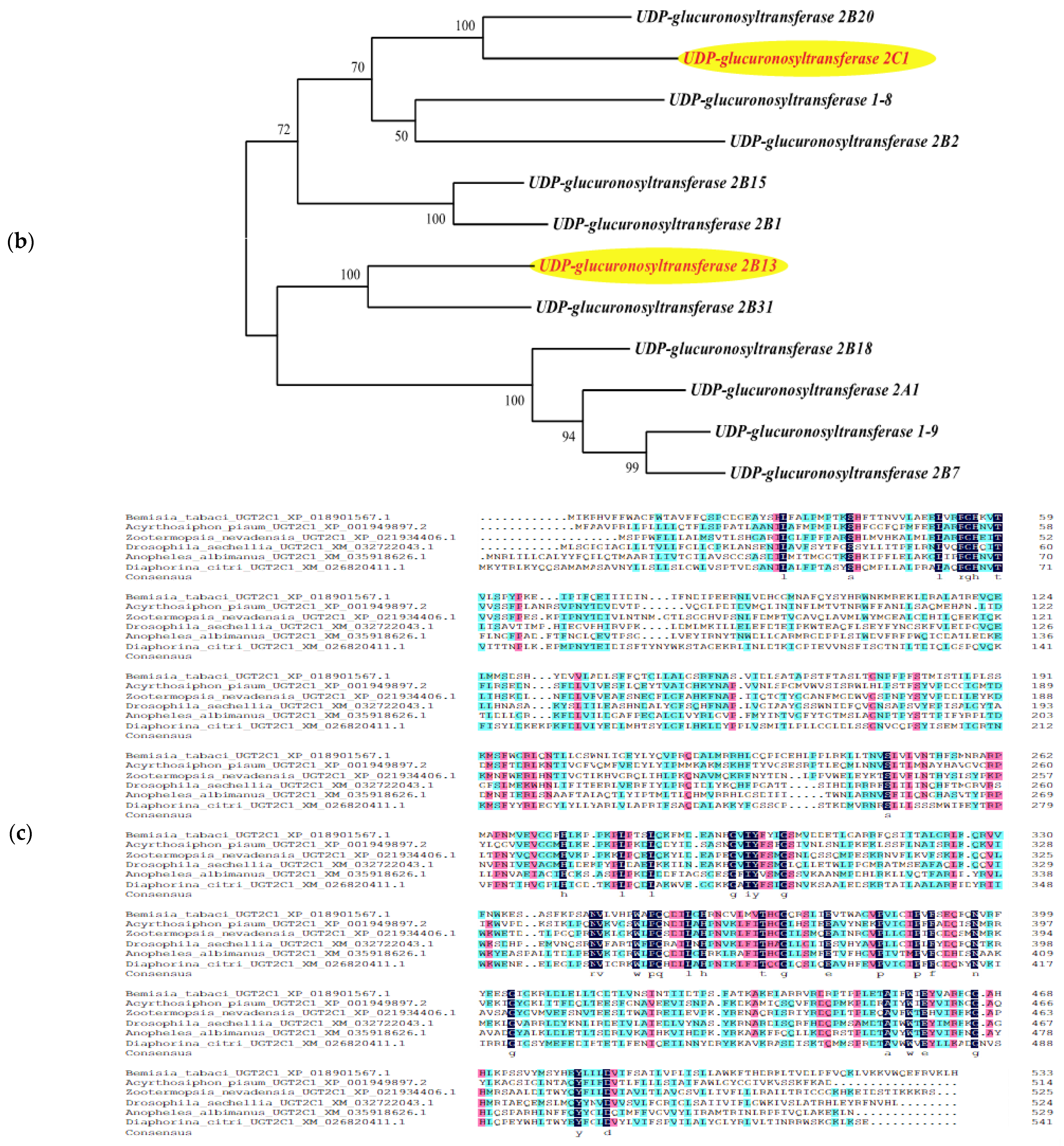
3.5. Bioinformatics Analysis of Genes from a Target Regulatory Pathway in B. tabaci
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Barro, P.J.; Liu, S.S.; Boykin, L.M.; Dinsdale, A.B. Bemisia tabaci: A statement of species status. Annu. Rev. Entomol. 2011, 56, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Dinsdale, A.; Cook, L.; Riginos, C.; Buckley, Y.M.; De Barro, P.D. Refined global analysis of Bemisia tabaci (Hemiptera: Sternorrhyncha: Aleyrodoidea: Aleyrodidae) mitochondrial cytochrome oxidase 1 to identify species level genetic boundaries. Ann. Entomol. Soc. Am. 2010, 103, 196–208. [Google Scholar] [CrossRef]
- Chu, D.; Zhang, Y.J.; Cong, B.; Xu, B.Y.; Wu, Q.J. Identification for Yunnan q-biotype population. Entomol. Knowl. 2005, 42, 54–56. [Google Scholar]
- Hu, J.; De Barro, P.; Zhao, H.; Wang, J.; Nardi, F.; Liu, S.S. An extensive field survey combined with a phylogenetic analysis reveals rapid and widespread invasion of two alien whiteflies in China. PLoS ONE 2011, 6, e16061. [Google Scholar] [CrossRef]
- Xue, Y.; Lin, C.; Wang, Y.; Zhang, Y.; Ji, L. Ecological niche complexity of invasive and native cryptic species of the bemisia tabaci species complex in china. J. Pest Sci. 2022, 95, 1245–1259. [Google Scholar] [CrossRef]
- Li, Z.X. Molecular phylogenetic analysis reveals at least five genetic races of Bemisia tabaci, in China. Phytoparasitica 2006, 34, 431–440. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Liu, S.S. Taxonomic status and distribution of Bemisia tabaci in China. J. Biosaf. 2012, 21, 247–255. [Google Scholar]
- Ma, F.Z.; Lü, Z.C.; Wang, R.; Wan, F.H. Heritability and evolutionary potential in thermal tolerance traits in the invasive Mediterranean cryptic species of Bemisia tabaci (Hemiptera: Aleyrodidae). PLoS ONE 2014, 9, e103279. [Google Scholar] [CrossRef]
- Shen, X.N.; Ji, S.X.; Liu, W.X.; Guo, J.Y.; Lü, Z.C.; Wan, F.H. Molecular characteristics of three cold resistance genes and their roles in temperature stress response in two Bemisia tabaci cryptic species. Entomol. Gen. 2021, 41, 317–328. [Google Scholar] [CrossRef]
- Storey, K.B.; Storey, J.M. Insect cold hardiness: Metabolic, gene, and protein adaptation. Can. J. Zool. 2012, 90, 456–475. [Google Scholar] [CrossRef]
- Liu, Y.C.; Su, H.; Li, R.Q.; Li, X.T.; Xu, Y.S.; Dai, X.P.; Zhou, Y.Y.; Wang, H.B. Comparative transcriptome analysis of Glyphodes pyloalis Walker (Lepidoptera: Pyralidae) reveals novel insights into heat stress tolerance in insects. BMC Genom. 2017, 18, 974. [Google Scholar] [CrossRef]
- Kang, Z.W.; Liu, F.H.; Liu, X.; Yu, W.B.; Tan, X.L.; Zhang, S.Z.; Tian, H.G.; Liu, T.X. The potential coordination of the heat-shock proteins and antioxidant enzyme genesof Aphidius gifuensis in response to thermal stress. Front. Physiol. 2017, 8, 976. [Google Scholar] [CrossRef]
- Chen, K.; Tang, T.; Song, Q.; Wang, Z.; He, K.; Liu, X.; Song, J.; Wang, L.; Yang, Y.; Feng, C. Transcription Analysis of the Stress and Immune Response Genes to Temperature Stress in Ostrinia furnacalis. Front. Physiol. 2019, 10, 1289. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, X.Q.; Xiao, P.A.; Tang, G.H.; Liu, S.H.; Lou, B.H.; Wang, J.J.; Jiang, H.B. Comparative transcriptome analysis reveals differentially expressed genes in the Asian citrus psyllid (Diaphorina citri) upon heat shock. Comp. Biochem. Physiol. Part D Genom. Proteom. 2019, 30, 256–261. [Google Scholar] [CrossRef]
- Catalán, T.P.; Wozniak, A.; Niemeyer, H.M.; Kalergis, A.M.; Bozinovic, F. Interplay between thermal and immune ecology: Effect of environmental temperature on insect immune response and energetic costs after an immune challenge. J. Insect Physiol. 2012, 58, 310–317. [Google Scholar] [CrossRef]
- Cui, M.; Hu, P.; Wang, T.; Tao, J.; Zong, S. Differential transcriptome analysis reveals genes related to cold tolerance in seabuckthorn carpenter moth, Eogystia hippophaecolus. PLoS ONE 2017, 12, e0187105. [Google Scholar] [CrossRef]
- Chang, Y.W.; Zhang, X.X.; Lu, M.X.; Gong, W.R.; Du, Y.Z. Transcriptome analysis of Liriomyza trifolii (Diptera: Agromyzidae) in response to temperature stress. Comp. Biochem. Physiol. Part D Genom. Proteom. 2020, 34, 100677. [Google Scholar] [CrossRef]
- Dunning, L.T.; Dennis, A.B.; Sinclair, B.J.; Newcomb, R.D.; Buckley, T.R. Divergent transcriptional responses to low temperature among populations of alpine and lowland species of New Zealand stick insects (Micrarchus). Mol. Ecol. 2014, 23, 2712–2726. [Google Scholar] [CrossRef]
- Hoffmann, A.A. Rapid adaptation of invertebrate pests to climatic stress? Curr. Opin. Insect. Sci. 2017, 21, 7–13. [Google Scholar] [CrossRef]
- Altschul, S.; Demchak, B.; Durbin, R.; Gentleman, R.; Krzywinski, M.; Li, H.; Nekrutenko, A.; Robinson, J.; Rasband, W.; Taylor, J.; et al. The anatomy of successful computational biology software. Nat. Biotechnol. 2013, 31, 894–897. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Chen, C.H.; Yang, Z.Z.; Guo, L.T.; Yang, X.; Wang, D.; Chen, M.; Huang, J.; Wen, Y.; Zeng, Y.; et al. Genome sequencing of the sweet potato whitefly Bemisia tabaci MED/Q. Gigascience 2017, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Allain, E.P.; Rouleau, M.; Lévesque, E.; Guillemette, C. Emerging roles for UDP-glucuronosyltransferases in drug resistance and cancer progression. Br. J. Cancer 2020, 122, 1277–1287. [Google Scholar] [CrossRef]
- Huang, X.; Ma, J.; Qin, X.; Tu, X.; Cao, G.; Wang, G.; Nong, X.; Zhang, Z. Biology, physiology and gene expression of grasshopper Oedaleus asiaticus exposed to diet stress from plant secondary compounds. Sci. Rep. 2017, 7, 8655. [Google Scholar] [CrossRef]
- Jurica, M.S.; Licklider, L.J.; Gygi, S.R.; Grigorieff, N.; Moore, M.J. Purification and characterization of native spliceosomes suitable for three-dimensional structural analysis. RNA 2002, 8, 426–439. [Google Scholar] [CrossRef]
- Wood, J.G.; Jones, B.C.; Jiang, N.; Chang, C.; Hosier, S.; Wickremesinghe, P.; Garcia, M.; Hartnett, D.A.; Burhenn, L.; Neretti, N.; et al. Chromatin-modifying genetic interventions suppress age-associated transposable element activation and extend life span in drosophila. Proc. Natl. Acad. Sci. USA 2016, 113, 11277–11282. [Google Scholar] [CrossRef]
- Qing, Y.; Yin, F.; Wang, W.; Zheng, Y.; Guo, P.; Schozer, F.; Deng, H.; Pan, D. The Hippo effector Yorkie activates transcription by interacting with a histone methyltransferase complex through Ncoa6. eLife 2014, 3, e02564. [Google Scholar] [CrossRef]
- Ji, S.X.; Wang, X.D.; Shen, X.N.; Liang, L.; Liu, W.X.; Wan, F.H.; Lü, Z.C. Using RNA interference to reveal the function of chromatin remodeling factor ISWI in temperature tolerance in Bemisia tabaci Middle East-Asia Minor 1 cryptic species. Insects 2020, 11, 113. [Google Scholar] [CrossRef]
- Patana, A.S.; Kurkela, M.; Finel, M.; Goldman, A. Mutation analysis in UGT1A9 suggests a relationship between substrate and catalytic residues in UDP-glucuronosyltransferases. Protein Eng. Des. Sel. 2008, 21, 537–543. [Google Scholar] [CrossRef]
- Bowles, D.; Isayenkova, J.; Lim, E.K.; Poppenberger, B. Glycosyltransferases: Managers of small molecules. Curr. Opin. Plant Biol. 2005, 8, 254–263. [Google Scholar] [CrossRef]
- Gachon, C.M.; Langlois-Meurinne, M.; Saindrenan, P. Plant secondary metabolism glycosyltransferases: The emerging functional analysis. Trends Plant Sci. 2005, 10, 542–549. [Google Scholar] [CrossRef]
- Miyauchi, Y.; Tanaka, Y.; Nagata, K.; Yamazoe, Y.; Mackenzie, P.I.; Yamada, H.; Ishii, Y. UDP-glucuronosyltransferase (UGT)-mediated attenuations of cytochrome P450 3A4 activity: UGT isoform-dependent mechanism of suppression. Br. J. Pharmacol. 2020, 177, 1077–1089. [Google Scholar] [CrossRef]
- Watts, M.; Pankhurst, N.W.; King, H.R. Maintenance of Atlantic salmon (Salmo salar) at elevated temperature inhibits cytochrome P450 aromatase activity in isolated ovarian follicles. Gen. Comp. Endocrinol. 2004, 135, 381–390. [Google Scholar] [CrossRef]
- Kumar, S.; Sun, L.; Liu, H.; Muralidhara, B.K.; Halpert, J.R. Engineering mammalian cytochrome P450 2b1 by directed evolution for enhanced catalytic tolerance to temperature and dimethyl sulfoxide. Protein Eng. Des. Sel. 2006, 19, 547–554. [Google Scholar] [CrossRef]
- Zhou, X.R.; Shan, Y.M.; Tan, Y.; Zhang, Z.R.; Pang, B.P. Comparative analysis of transcriptome responses to cold stress in galeruca daurica (Coleoptera: Chrysomelidae). J. Insect Sci. 2019, 19, 1–6. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, M.; Liu, Y.; Zhou, Z.; Guo, J. Identification of cytochrome P450 monooxygenase genes and their expression in response to high temperature in the alligatorweed flea beetle Agasicles hygrophila (Coleoptera: Chrysomelidae). Sci. Rep. 2018, 8, 17847. [Google Scholar] [CrossRef]
- Kang, J.; Kim, J.; Choi, K.W. Novel cytochrome P450, cyp6a17, is required for temperature preference behavior in drosophila. PLoS ONE 2011, 6, e29800. [Google Scholar] [CrossRef]
- Shen, X.N.; Liu, W.X.; Wan, F.H.; Lv, Z.C.; Guo, J.Y. The Role of cytochrome P450 4C1 and carbonic anhydrase 3 in Response to temperature stress in Bemisia tabaci. Insects 2021, 12, 1071. [Google Scholar] [CrossRef]
- Gautam, S.; Crossley, M.S.; Dutta, B.; Coolong, T.; Simmons, A.M.; da Silva, A.; Snyder, W.E.; Srinivasan, R. Low Genetic Variability in Bemisia tabaci MEAM1 Populations within Farmscapes of Georgia, USA. Insects 2020, 11, 834. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviated Name | Full Gene Name | Amino Acids | T vs. H | N vs. H | Putative Functions (from NCBI Database) |
|---|---|---|---|---|---|
| BtADRO | adrenodoxin oxidoreductase | 459 | up | up | |
| BtUraD | 2-oxo-4-hydroxy-4-carboxy-5-ureidoimidazoline decarboxylase | 174 | |||
| BtUGT2B13 | UDP-glucuronosyltransferase 2B13 | 522 | |||
| BtMet16 | U6 small nuclear RNA (adenine-(43)-N(6))-methyltransferase | 524 | involved in regulation of mRNA splicing via spliceosome | ||
| Btmsps | Protein mini spindles | 1997 | |||
| BtSPF27 | Pre-mRNA-splicing factor SPF27 | 221 | activates pre-mRNA splicing | ||
| BtCacybp | Calcyclin-binding protein | 224 | |||
| BtMal1 | Maltase 1 | 600 | maltose alpha-glucosidase activity | ||
| BtOzf6 | Oocyte zinc finger protein XlCOF6 | 381 | trypsin-like serine protease activity | ||
| BtUbp5 | Ubiquitin carboxyl-terminal hydrolase 5 | 813 | |||
| BtBOLA3 | BolA-like protein 3 | 104 | |||
| BtF10C1 | Protein FRA10AC1 homolog | 249 | involved in dephosphorylation | ||
| BtALG13 | UDP-N-acetylglucosamine transferase and deubiquitinase ALG13 | 745 | involved in proteolysis | ||
| BtSpin | Protein spinster | 531 | down | down | enables transmembrane transporter activity |
| BtUGT2C1 | UDP-glucuronosyltransferase 2C1 | 534 | |||
| BtAnnu | Annulin | 756 | |||
| BtSiah2 | E3 ubiquitin-protein ligase siah2 | 155 | involved in apoptotic process | ||
| BtTrePt | Transposable element P transposase | 1157 | |||
| BtKAE1 | tRNA N6-adenosine threonylcarbamoyltransferase | 334 | involved in chromosome organization, enables chromatin DNA binding |
| Abbreviated Name | Amino Acids | Signal Peptide | Transmembrane Region | Pfam: UDPGT | Pfam: Glyco_tran_28_C | NCBI Reference Sequence ID |
|---|---|---|---|---|---|---|
| BtUGT1-8 | 533 | 1–19 | 474–496 | 31–517 | (-) | XP_018907955.1 |
| BtUGT1-9 | 527 | (-) | 7–29, 492–514 | 21–524 | (-) | XP_018897720.1 |
| BtUGT2A1 | 528 | 1–19 | 390–409 | 21–527 | 330–457 | XP_018909896.1 |
| BtUGT2B1 | 518 | (-) | 7–29, 480–502 | 25–515 | 303–444 | XP_018896780.1 |
| BtUGT2B2 | 519 | 1–19 | 473–495 | 22–514 | (-) | XP_018899270.1 |
| BtUGT2B7 | 527 | (-) | 7–29, 480–508 | 21–523 | (-) | XP_018896827.1 |
| BtUGT2B13 | 522 | 1–19 | 479–501 | 21–515 | 304–444 | XP_018905849.1 |
| BtUGT2B15 | 518 | 1–22 | 480–502 | 24–517 | 480–502 | XP_018896916.1 |
| BtUGT2B18 | 537 | (-) | 7–29, 494–516 | 26–534 | 340–469 | XP_018912485.1 |
| BtUGT2B20 | 526 | (-) | 12–34, 488–510 | 31–525 | (-) | XP_018911460.1 |
| BtUGT2B31 | 512 | 1–20 | 480–502 | 21–504 | (-) | XP_018905835.1 |
| BtUGT2C1 | 534 | 1–26 | 482–504 | 29–510 | 302–446 | XP_018901567.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, X.-N.; Wang, X.-D.; Wan, F.-H.; Lü, Z.-C.; Liu, W.-X. Gene Expression Analysis Reveals Potential Regulatory Factors Response to Temperature Stress in Bemisia tabaci Mediterranean. Genes 2023, 14, 1013. https://doi.org/10.3390/genes14051013
Shen X-N, Wang X-D, Wan F-H, Lü Z-C, Liu W-X. Gene Expression Analysis Reveals Potential Regulatory Factors Response to Temperature Stress in Bemisia tabaci Mediterranean. Genes. 2023; 14(5):1013. https://doi.org/10.3390/genes14051013
Chicago/Turabian StyleShen, Xiao-Na, Xiao-Di Wang, Fang-Hao Wan, Zhi-Chuang Lü, and Wan-Xue Liu. 2023. "Gene Expression Analysis Reveals Potential Regulatory Factors Response to Temperature Stress in Bemisia tabaci Mediterranean" Genes 14, no. 5: 1013. https://doi.org/10.3390/genes14051013
APA StyleShen, X.-N., Wang, X.-D., Wan, F.-H., Lü, Z.-C., & Liu, W.-X. (2023). Gene Expression Analysis Reveals Potential Regulatory Factors Response to Temperature Stress in Bemisia tabaci Mediterranean. Genes, 14(5), 1013. https://doi.org/10.3390/genes14051013