

Familial 4p Interstitial Deletion Provides New Insights and Candidate Genes Underlying This Rare Condition



Abstract
1. Introduction
2. Case Presentation
3. Methods and Results
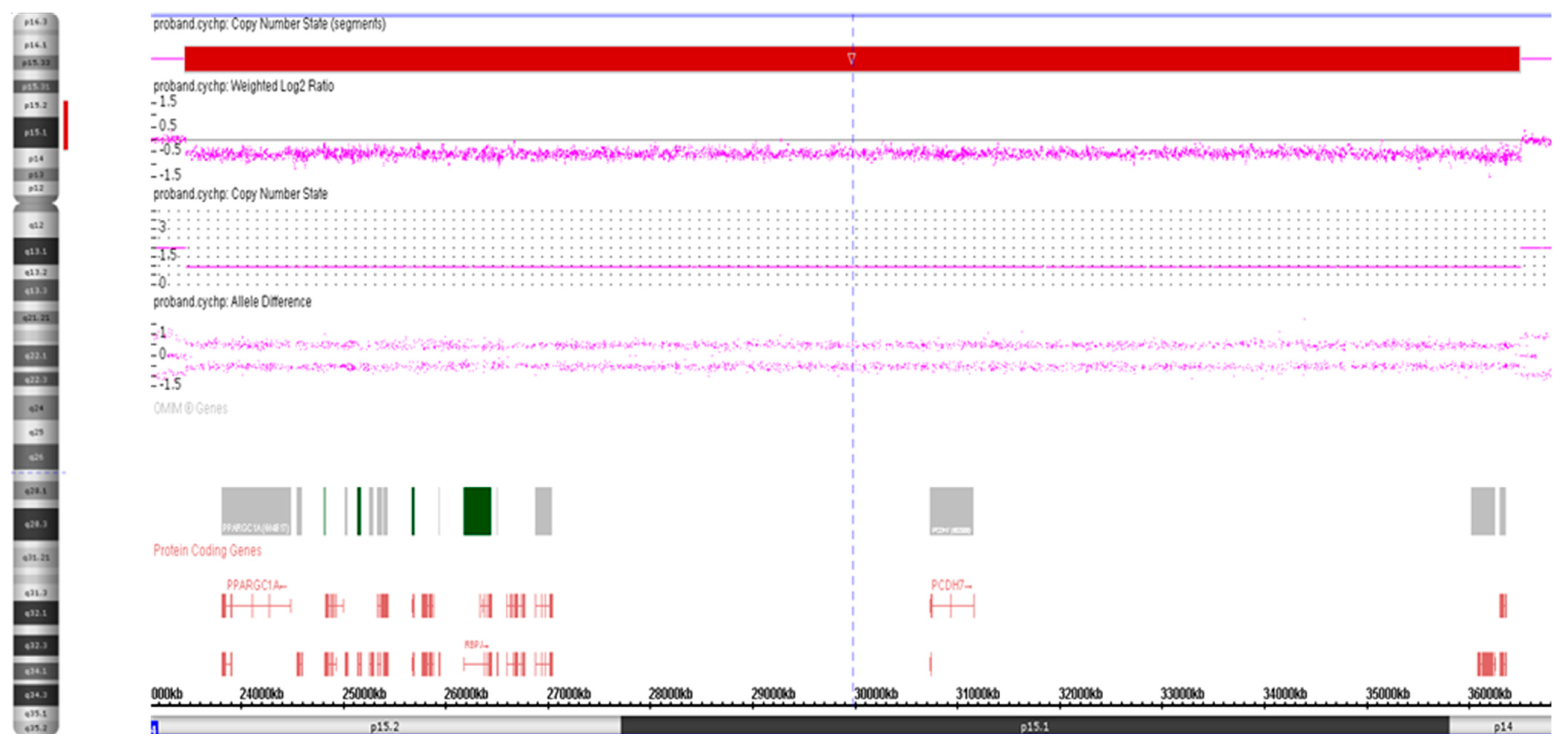
3.1. Cytogenetic Analysis
3.2. High-Resolution SNP Array Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Altherr, M.R.; Bengtsson, U.; Elder, F.F.; Ledbetter, D.H.; Wasmuth, J.J.; McDonald, M.E.; Gusella, J.F.; Greenberg, F. Molecular confirmation of Wolf-Hirschhorn syndrome with a subtle translocation of chromosome 4. Am. J. Hum. Genet. 1991, 49, 1235–1242. [Google Scholar]
- Alesi, V.; Barrano, G.; Morara, S.; Darelli, D.; Petrilli, K.; Capalbo, A.; Pacella, M.; Haass, C.; Finocchi, M.; Novelli, A.; et al. A previously undescribed de novo 4p15 deletion in a patient with apparently isolated metopic craniosynostosis. Am. J. Med. Genet. A 2011, 155A, 2543–2551. [Google Scholar] [CrossRef]
- Basinko, A.; Douet-Guilbert, N.; Parent, P.; Blondin, G.; Mingam, M.; Monot, F.; Morel, F.; Le Bris, M.-J.; De Braekeleer, M. Familial interstitial deletion of the short arm of chromosome 4 (p15.33–p16.3) characterized by molecular cytogenetic analysis. Am. J. Med. Genet. Part A 2008, 146A, 899–903. [Google Scholar] [CrossRef]
- Estabrooks, L.L.; Rao, K.W.; Korf, B. Interstitial deletion of distal chromosome 4p in a patient without classical Wolf-Hirschhorn syndrome. Am. J. Med. Genet. 1993, 45, 97–100. [Google Scholar] [CrossRef] [PubMed]
- White, D.M.; Pillers, D.-A.M.; Reiss, J.A.; Brown, M.G.; Magenis, R.E. Interstitial deletions of the short arm of chromosome 4 in patients with a similar combination of multiple minor anomalies and mental retardation. Am. J. Med. Genet. 1995, 57, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Van De Graaf, G.; Sijstermans, J.M.; Engelen, J.J.; Schrander-Stumpel, C.T. Mild phenotype in interstitial 4p deletion: Another patient and review of the literature. Genet. Couns. 1997, 8, 13–18. [Google Scholar] [PubMed]
- Tonk, V.S.; Jalal, S.M.; Gonzalez, J.; Kennedy, A.; Velagaleti, G.V. Familial interstitial deletion of chromosome 4 (p15.2p16.1). Ann. Genet. 2003, 46, 453–458. [Google Scholar] [CrossRef]
- Su, P.-H.; Lee, I.-C.; Chen, J.-Y.; Chen, S.-J.; Yu, J.-S.; Tsao, T.-F. Interstitial Deletions of the Short Arm of Chromosome 4 in a Patient With Mental Retardation and Focal Seizure. Pediatr. Neonatol. 2011, 52, 165–168. [Google Scholar] [CrossRef]
- South, S.T.; Corson, V.L.; McMichael, J.L.; Blakemore, K.J.; Stetten, G. Prenatal Detection of an Interstitial Deletion in 4p15 in a Fetus with an Increased Nuchal Skin Fold Measurement. Fetal Diagn. Ther. 2004, 20, 58–63. [Google Scholar] [CrossRef]
- Ray, M.; Evans, J.; Rockman-Greenberg, C.; Wickstrom, D. Interstitial deletion of the short arm of chromosome 4. J. Med. Genet. 1984, 21, 223–225. [Google Scholar] [CrossRef]
- Piovani, G.; Borsani, G.; Bertini, V.; Kalscheuer, V.M.; Viertel, P.; Bellotti, D.; Valseriati, D.; Barlati, S. Unexpected identification of two interstitial deletions in a patient with a pericentric inversion of a chromosome 4 and an abnormal phenotype. Eur. J. Med. Genet. 2006, 49, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Møller, R.; Hansen, C.; Ullmann, R.; Ropers, H.; Tommerup, N.; Tümer, Z.; Jackson, G. Interstitial deletion of chromosome 4p associated with mild mental retardation, epilepsy and polymicrogyria of the left temporal lobe. Clin. Genet. 2007, 72, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Mitroi, A.; Aschie, M.; Apostol, A.; Brinzan, C.; Cozaru, G.; Mitroi, A.N. A boy with 13.34-Mb interstitial deletion of chromosome 4p15: A new case report and review of the literature. Medicine 2017, 96, e9301. [Google Scholar] [CrossRef]
- Ishikawa, T.; Sumi, S.; Fujimoto, S.; Shima, Y.; Wada, Y. Interstitial deletion of the short arm of chromosome 4 in a boy with mild psychomotor retardation and dysmorphism. Clin. Genet. 1990, 38, 314–317. [Google Scholar] [CrossRef]
- Fryns, J.P.; Yang-Aisheng; Kleczkowska, A.; Lemmens, F.; Vandecasseye, W.; van den Berghe, H. Interstitial deletion of the short arm of chromosome 4. A phenotype distinct from the Wolf-Hirschhorn syndrome. Ann. Genet. 1989, 32, 59–61. [Google Scholar] [PubMed]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Jeon, B.R.; Lee, Y.K.; Ki, C.-S.; Jang, M.-A. The First Korean Case of De Novo Proximal 4p Deletion Syndrome in a Child With Developmental Delay. Ann. Lab. Med. 2020, 40, 435–437. [Google Scholar] [CrossRef] [PubMed]
- Krebs, L.T.; Shutter, J.R.; Tanigaki, K.; Honjo, T.; Stark, K.L.; Gridley, T. Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev. 2004, 18, 2469–2473. [Google Scholar] [CrossRef]
- Giaimo, B.D.; Gagliani, E.K.; Kovall, R.A.; Borggrefe, T. Transcription Factor RBPJ as a Molecular Switch in Regulating the Notch Response. Adv. Exp. Med. Biol. 2020, 1287, 9–30. [Google Scholar] [CrossRef]
- Shen, W.; Huang, J.; Wang, Y. Biological Significance of NOTCH Signaling Strength. Front. Cell Dev. Biol. 2021, 9, 652273. [Google Scholar] [CrossRef]
- Blackwood, C.A.; Bailetti, A.; Nandi, S.; Gridley, T.; Hébert, J.M. Notch Dosage: Jagged1 Haploinsufficiency Is Associated With Reduced Neuronal Division and Disruption of Periglomerular Interneurons in Mice. Front. Cell Dev. Biol. 2020, 8, 113. [Google Scholar] [CrossRef]
- McCright, B.; Lozier, J.; Gridley, T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development 2002, 129, 1075–1082. [Google Scholar] [CrossRef]
- Hassed, S.J.; Wiley, G.B.; Wang, S.; Lee, J.-Y.; Li, S.; Xu, W.; Zhao, Z.J.; Mulvihill, J.J.; Robertson, J.; Warner, J.; et al. RBPJ Mutations Identified in Two Families Affected by Adams-Oliver Syndrome. Am. J. Hum. Genet. 2012, 91, 391–395. [Google Scholar] [CrossRef]
- Dong, Y.; Jesse, A.M.; Kohn, A.; Gunnell, L.M.; Honjo, T.; Zuscik, M.; O'Keefe, R.J.; Hilton, M.J. RBPjκ-dependent Notch signaling regulates mesenchymal progenitor cell proliferation and differentiation during skeletal development. Development 2010, 137, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Vauclair, S.; Nicolas, M.; Barrandon, Y.; Radtke, F. Notch1 is essential for postnatal hair follicle development and homeostasis. Dev. Biol. 2005, 284, 184–193. [Google Scholar] [CrossRef]
- Dou, G.; Wang, Y.; Hu, X.; Hou, L.; Wang, C.; Xu, J.; Wang, Y.; Liang, Y.; Yao, L.; Yang, A.; et al. RBP-J, the transcription factor downstream of Notch receptors, is essential for the maintenance of vascular homeostasis in adult mice. FASEB J. 2007, 22, 1606–1617. [Google Scholar] [CrossRef]
- Nakayama, T.; Saitsu, H.; Endo, W.; Kikuchi, A.; Uematsu, M.; Haginoya, K.; Hino-Fukuyo, N.; Kobayashi, T.; Iwasaki, M.; Tominaga, T.; et al. RBPJ is disrupted in a case of proximal 4p deletion syndrome with epilepsy. Brain Dev. 2013, 36, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Soboloff, J.; Spassova, M.A.; Tang, X.D.; Hewavitharana, T.; Xu, W.; Gill, D.L. Orai1 and STIM Reconstitute Store-operated Calcium Channel Function. J. Biol. Chem. 2006, 281, 20661–20665. [Google Scholar] [CrossRef] [PubMed]
- Mancarella, S.; Potireddy, S.; Wang, Y.; Gao, H.; Gandhirajan, R.K.; Autieri, M.; Scalia, R.; Cheng, Z.; Wang, H.; Madesh, M.; et al. Targeted STIM deletion impairs calcium homeostasis, NFAT activation, and growth of smooth muscle. FASEB J. 2012, 27, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.T.; Alevizos, I.; Liu, X.; Swaim, W.D.; Yin, H.; Feske, S.; Oh-Hora, M.; Ambudkar, I.S. STIM1 and STIM2 protein deficiency in T lymphocytes underlies development of the exocrine gland autoimmune disease, Sjögren’s syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 14544–14549. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, L.S.; Graham, S.J.L.; Dziadek, M.A. STIM proteins: Integrators of signalling pathways in development, differentiation and disease. J. Cell. Mol. Med. 2010, 14, 1890–1903. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; McCarl, C.-A.; Papolos, A.; Khalil, S.; Lüthy, K.; Hivroz, C.; LeDeist, F.; Rieux-Laucat, F.; Rechavi, G.; Rao, A.; et al. STIM1 Mutation Associated with a Syndrome of Immunodeficiency and Autoimmunity. N. Engl. J. Med. 2009, 360, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Oh-Hora, M.; Yamashita, M.; Hogan, P.G.; Sharma, S.; Lamperti, E.; Chung, W.; Prakriya, M.; Feske, S.; Rao, A. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat. Immunol. 2008, 9, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Popugaeva, E.; Pchitskaya, E.; Speshilova, A.; Alexandrov, S.; Zhang, H.; Vlasova, O.; Bezprozvanny, I. STIM2 protects hippocampal mushroom spines from amyloid synaptotoxicity. Mol. Neurodegener. 2015, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Zhang, Q.; Wu, Z.; Ma, T.; He, A.; Zhang, T.; Ke, X.; Yu, Q.; Han, Y.; Lu, Y. Mossy cell synaptic dysfunction causes memory imprecision via miR-128 inhibition of STIM2 in Alzheimer's disease mouse model. Aging Cell 2020, 19, e13144. [Google Scholar] [CrossRef]
- Serwach, K.; Gruszczynska-Biegala, J. Target Molecules of STIM Proteins in the Central Nervous System. Front. Mol. Neurosci. 2020, 13, 617422. [Google Scholar] [CrossRef]
- Cheng, H.; Chen, L.; Fang, Z.; Wan, Q.; Du, Z.; Ma, N.; Guo, G.; Lu, W. STIM2 promotes the invasion and metastasis of breast cancer cells through the NFAT1/TGF-β1 pathway. Cell. Mol. Biol. 2022, 67, 55–61. [Google Scholar] [CrossRef]
- Fleur-Lominy, S.S.; Maus, M.; Vaeth, M.; Lange, I.; Zee, I.; Suh, D.; Liu, C.; Wu, X.; Tikhonova, A.; Aifantis, I.; et al. STIM1 and STIM2 Mediate Cancer-Induced Inflammation in T Cell Acute Lymphoblastic Leukemia. Cell Rep. 2018, 24, 3045–3060.e5. [Google Scholar] [CrossRef]
- Abdelhaleem, M.; Maltais, L.; Wain, H. The human DDX and DHX gene families of putative RNA helicases. Genomics 2003, 81, 618–622. [Google Scholar] [CrossRef]
- Imamura, O.; Sugawara, M.; Furuichi, Y. Cloning and Characterization of a Putative Human RNA Helicase Gene of the DEAH-Box Protein Family. Biochem. Biophys. Res. Commun. 1997, 240, 335–340. [Google Scholar] [CrossRef]
- Wang, P.; Zhu, S.; Yang, L.; Cui, S.; Pan, W.; Jackson, R.; Zheng, Y.; Rongvaux, A.; Sun, Q.; Yang, G.; et al. Nlrp6 regulates intestinal antiviral innate immunity. Science 2015, 350, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Nakano, K.; Shimizu, T.; Ohto, U. The crystal structure of human DEAH-box RNA helicase 15 reveals a domain organization of the mammalian DEAH/RHA family. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2017, 73 Pt 6, 347–355. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, X.; Pan, L.; Huang, Y.; Cai, Y.; Li, J.; Li, Y.; Wang, S. RNA helicase DHX15 decreases cell apoptosis by NF-κB signaling pathway in Burkitt lymphoma. Cancer Cell Int. 2022, 22, 92. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Chen, K.; Qin, Y.; Niu, Y.; Zhang, X.; Xu, S.; Zhang, C.; Feng, M.; Wang, K. Long Non-coding RNA JHDM1D-AS1 Interacts with DHX15 Protein to Enhance Non-Small-Cell Lung Cancer Growth and Metastasis. Mol. Ther. Nucleic Acids 2019, 18, 831–840. [Google Scholar] [CrossRef]
- Faber, Z.J.; Chen, X.; Gedman, A.L.; Boggs, K.; Cheng, J.; Ma, J.; Radtke, I.; Chao, J.-R.; Walsh, M.P.; Song, G.; et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat. Genet. 2016, 48, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Li, Y.; Zhang, H.-Y.; Zheng, Y.; Liu, X.-L.; Hu, Z.; Wang, Y.; Wang, J.; Cai, Y.-H.; Liu, Q.; et al. DHX15 is associated with poor prognosis in acute myeloid leukemia (AML) and regulates cell apoptosis via the NF-kB signaling pathway. Oncotarget 2017, 8, 89643–89654. [Google Scholar] [CrossRef]
- Liang, H.; Ward, W.F. PGC-1alpha: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef]
- Austin, S.; St-Pierre, J. PGC1α and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125, 4963–4971. [Google Scholar] [CrossRef]
- Blasiak, J.; Szczepanska, J.; Fila, M.; Pawlowska, E.; Kaarniranta, K. Potential of Telomerase in Age-Related Macular Degeneration-Involvement of Senescence, DNA Damage Response and Autophagy and a Key Role of PGC-1α. Int. J. Mol. Sci. 2021, 22, 7194. [Google Scholar] [CrossRef]
- Kuczynska, Z.; Metin, E.; Liput, M.; Buzanska, L. Covering the Role of PGC-1α in the Nervous System. Cells 2021, 11, 111. [Google Scholar] [CrossRef]
- Yoshida, K.; Yoshitomo-Nakagawa, K.; Seki, N.; Sasaki, M.; Sugano, S. Cloning, expression analysis, and chromosomal localization of BH-protocadherin (PCDH7), a novel member of the cadherin superfamily. Genomics 1998, 49, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Yasuda, S.; Tanaka, H.; Yamagata, K.; Kim, H. Non-clustered protocadherin. Cell Adhes. Migr. 2011, 5, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Heggem, M.A.; Bradley, R.S. The cytoplasmic domain of Xenopus NF-protocadherin interacts with TAF1/set. Dev. Cell 2003, 4, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Rashid, D.; Newell, K.; Shama, L.; Bradley, R. A requirement for NF-protocadherin and TAF1/Set in cell adhesion and neural tube formation. Dev. Biol. 2006, 291, 170–181. [Google Scholar] [CrossRef]
- Yoshida, K. Fibroblast cell shape and adhesion in vitro is altered by overexpression of the 7a and 7b isoforms of protocadherin 7, but not the 7c isoform. Cell Mol. Biol. Lett. 2003, 8, 735–741. [Google Scholar]
- Miyake, K.; Hirasawa, T.; Soutome, M.; Itoh, M.; Goto, Y.I.; Endoh, K.; Takahashi, K.; Kudo, S.; Nakagawa, T.; Yokoi, S.; et al. The protocadherins, PCDHB1 and PCDH7, are regulated by MeCP2 in neuronal cells and brain tissues: Implication for pathogenesis of Rett syndrome. BMC Neurosci. 2011, 12, 81. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | OMIM | Protein/Transcript Name | Function/Dysfunction of Gene Product |
|---|---|---|---|
| PPARGC1A | 604,517 | Peroxisome proliferative activated receptor γ | Transcriptional coactivator for steroid receptors and nuclear receptors |
| DHX15 | 603,403 | DEAH box polypeptide 15 | Nuclear ATP-dependent helicase |
| SOD3 | 185,490 | Superoxide dismutase 3 | Free radical detoxification |
| LGI2 | 608,301 | Leucine-rich glioma inactivated protein 2 | May be involved in axonal path finding |
| SEPSECS | 613,009 | O-phosphoserin tRNA-selenocystein tRNA synthase | Pontocerebellar hypoplasia type 2D (AR) |
| PI4K2B | 612,101 | Phosphatidylinositol 4-kinase type 2 β | Phosphatidylinositol 4-kinase type 2 β |
| ZCCHC4 | 611,792 | Zinc finger CCHC domain-containing protein 4 | May be a methyltransferase |
| ANAPC4 | 606,947 | Anaphase-promoting complex subunit 4 | Component of anaphase-promoting complex/cyclosome, a cell cycle-regulated E3 ubiquitin ligase and the G1 phase of the cell cycle |
| SLC34A2 | 604,217 | Sollute carrier family 34 (sodium, phosphate cotransporter) member 2 | Testicular microlithiasis. Pulmonary alveolar microlithiasisy (AR) |
| RBPJ | 147,183 | Recombination signal-binding protein for kappa J region | Adam-Oliver syndrome 3 (AD) |
| CCKAR | 118,444 | Colecystokinin A receptor | Receptor for cholecystokinin with role in colecystokinin induced regulation of satiety |
| PCDH7 | 602,988 | Procadherin 7 isoform c precursor | Mediation of calcium dependent cell-cell adhesion expressed predominantly in SNC |
| STIM2 | 610,841 | Stromal interaction molecule 2 | Regulation of basal cytosolic and endoplasmic reticulum Ca2+ concentrations |
| SMIM20 | 617,465 | Small Integral Membrane Protein 20 | Component of the MITRAC complex that regulates cytochrome C oxidase assembly |
| ARAP2 | 606,645 | Centaurin, Delta-1 | Phosphatidylinositol 3,4,5-trisphosphate-dependent GTPase-activating protein that modulates actin cytoskeleton remodeling by regulating ARF and RHO family members |
| DTHD1 | 616,979 | Death Domain-containing protein 1 | Death domain containing proteins function in signaling pathways and formation of signaling complexes, as well as the apoptosis pathway |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di, J.; Yenwongfai, L.; Rieger, H.T.; Zhang, S.; Wei, S. Familial 4p Interstitial Deletion Provides New Insights and Candidate Genes Underlying This Rare Condition. Genes 2023, 14, 635. https://doi.org/10.3390/genes14030635
Di J, Yenwongfai L, Rieger HT, Zhang S, Wei S. Familial 4p Interstitial Deletion Provides New Insights and Candidate Genes Underlying This Rare Condition. Genes. 2023; 14(3):635. https://doi.org/10.3390/genes14030635
Chicago/Turabian StyleDi, Jing, Leonard Yenwongfai, Hillary T. Rieger, Shulin Zhang, and Sainan Wei. 2023. "Familial 4p Interstitial Deletion Provides New Insights and Candidate Genes Underlying This Rare Condition" Genes 14, no. 3: 635. https://doi.org/10.3390/genes14030635
APA StyleDi, J., Yenwongfai, L., Rieger, H. T., Zhang, S., & Wei, S. (2023). Familial 4p Interstitial Deletion Provides New Insights and Candidate Genes Underlying This Rare Condition. Genes, 14(3), 635. https://doi.org/10.3390/genes14030635