
Hereditary Cancer Syndrome in a Family with Double Mutation in BRIP1 and MUTYH Genes

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
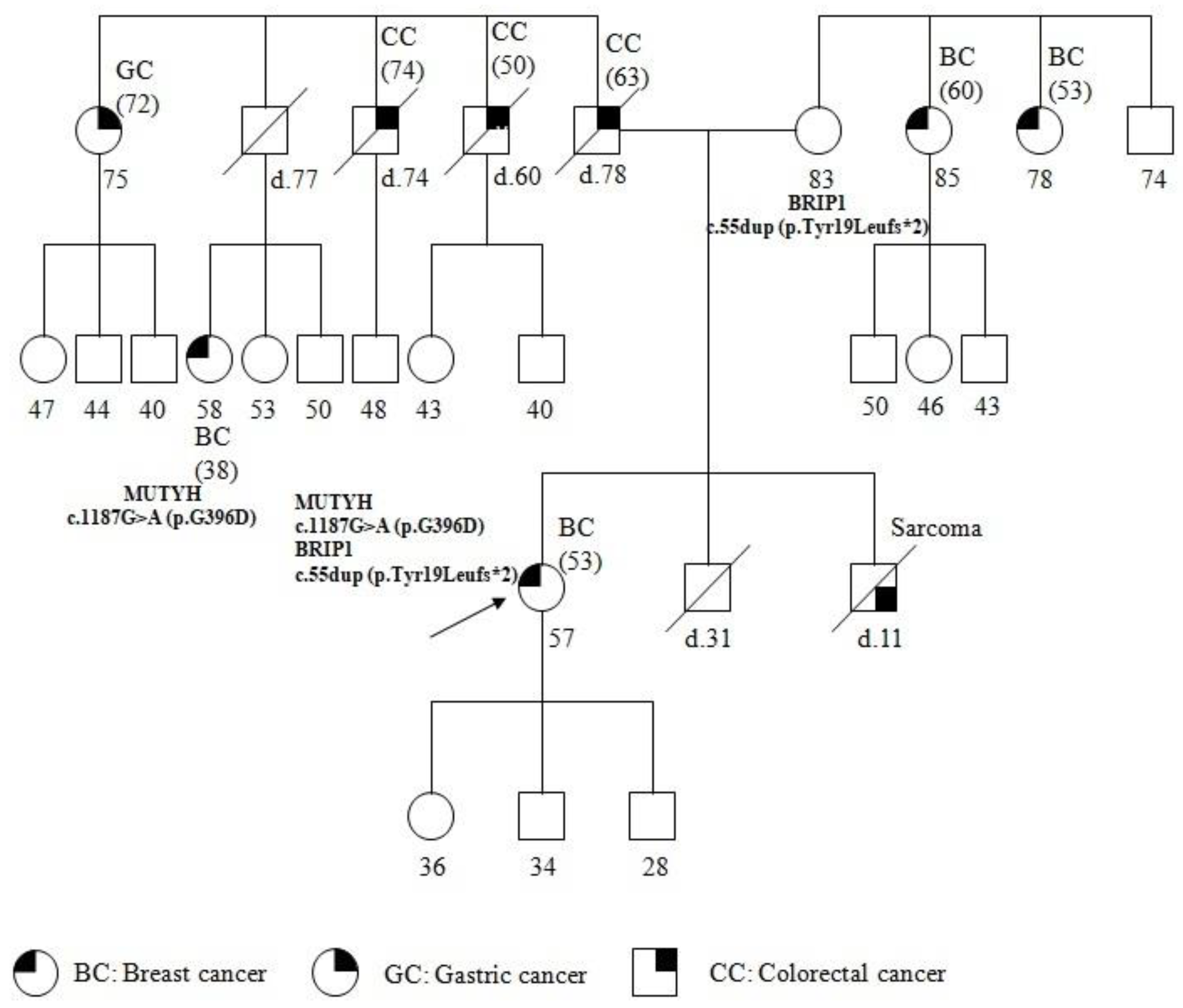
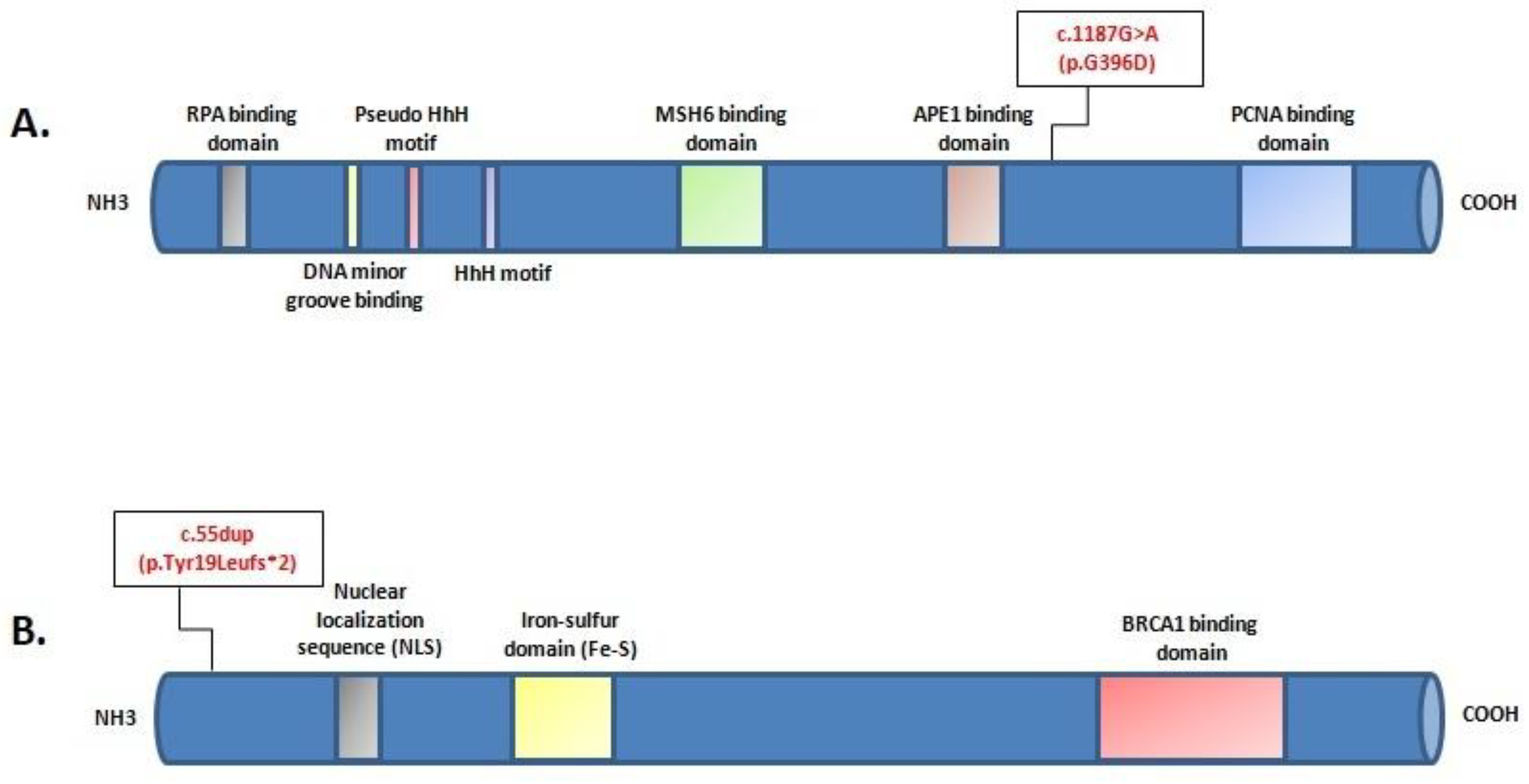
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American College of Obstetricians and Gynecologists. Hereditary Cancer Syndromes and Risk Assessment: ACOG COMMITTEE OPINION, Number 793. [No authors listed]. Obstet. Gynecol. 2019, 134, e143–e149. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.T.; D’Elia, G.; Caliendo, G.; Casamassimi, A.; Resse, M.; Passariello, L.; Cioffi, M.; Molinari, A.M. Double mutation of APC and BRCA1 in an Italian family. Cancer Genet. 2020, 244, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, H.; Yu, Z.; Li, L.; Zhang, J.; Liang, X.; Huang, Q. Germline variants profiling of BRCA1 and BRCA2 in Chinese Hakka breast and ovarian cancer patients. BMC Cancer 2022, 22, 842. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.T.; Caliendo, G.; D’Elia, G.; Resse, M.; Casamassimi, A.; Minucci, P.B.; DelloIoio, C.; Cioffi, M.; Molinari, A.M. Five Italian Families with Two Mutations in BRCA Genes. Genes 2020, 11, 1451. [Google Scholar] [CrossRef]
- Mazzei, F.; Viel, A.; Bignamia, M. Role of MUTYH in human cancer. Mutat. Res. 2013, 743–744, 33–43. [Google Scholar] [CrossRef]
- Talseth-Palmer, B.A.; Wijnen, J.T.; Andreassen, E.K.; Barker, D.; Jagmohan-Changur, S.; Tops, C.M.; Meldrum, C.; Dutch Cancer Genetics Group; Spigelman, A.; Hes, F.J.; et al. The importance of a large sample cohort for studies on modifier genes influencing disease severity in FAP patients. Hered. Cancer Clin. Pract. 2013, 11, 20. [Google Scholar] [CrossRef]
- D’Elia, G.; Caliendo, G.; Casamassimi, A.; Cioffi, M.; Molinari, A.M.; Vietri, M.T. APC and MUTYH Analysis in FAP Patients: A Novel Mutation in APC Gene and Genotype-Phenotype Correlation. Genes 2018, 9, 322. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, R.A.S.; Sabbaga, J.; Rossi, B.M.; Achatz, M.I.W.; Bettoni, F.; Camargo, A.A.; Asprino, P.F.; Galante, P.A.F. Monoallelic deleterious MUTYH germline variants as a driver for tumorigenesis. J. Pathol. 2022, 256, 214–222. [Google Scholar] [CrossRef]
- Curia, M.C.; Catalano, T.; Aceto, G.M. MUTYH: Not just polyposis. World J. Clin. Oncol. 2020, 11, 428–449. [Google Scholar] [CrossRef]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef]
- Yoshida, R. Hereditarybreast and ovariancancer (HBOC): Review of its molecular characteristics, screening, treatment, and prognosis. Breast Cancer 2021, 28, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Samuel, D.; Diaz-Barbe, A.; Pinto, A.; Schlumbrecht, M.; George, S. Hereditary Ovarian Carcinoma: Cancer Pathogenesis Looking beyond BRCA1 and BRCA2. Cells 2022, 11, 539. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.; Ghazali, M.B.; Wahab, M.A.M.A.; Yusoff, N.M.; Mahsin, H.; Seng, C.E.; Khalid, I.A.; Rahman, M.N.G.; Yahaya, B.H. The BRCA1 and BRCA2 Genes in Early-Onset Breast Cancer Patients. Adv. Exp. Med. Biol. 2020, 1292, 1–12. [Google Scholar] [CrossRef]
- Rafnar, T.; Gudbjartsson, D.F.; Sulem, P.; Jonasdottir, A.; Sigurdsson, A.; Jonasdottir, A.; Besenbacher, S.; Lundin, P.; Stacey, S.N.; Gudmundsson, J.; et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat. Genet. 2011, 43, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Guardiola, V.; Sarabia-Meseguer, M.D.; Marín-Vera, M.; Sánchez-Bermúdez, A.I.; Alonso-Romero, J.L.; Noguera-Velasco, J.A.; Ruiz-Espejo, F. New insights into the performance of multigene panel testing: Two novel nonsense variants in BRIP1 and TP53 in a young woman with breast cancer. Cancer Genet. 2018, 228–229, 1–4. [Google Scholar] [CrossRef]
- Gargallo, P.; Yáñez, Y.; Segura, V.; Juan, A.; Torres, B.; Balaguer, J.; Oltra, S.; Castel, V.; Cañete, A. Li-Fraumeni syndrome heterogeneity. Clin. Transl. Oncol. 2020, 22, 978–988. [Google Scholar] [CrossRef]
- Kamihara, J.; Rana, H.Q.; Garber, J.E. Germline TP53 Mutations and the Changing Landscape of Li-Fraumeni Syndrome. Hum. Mutat. 2014, 35, 654–662. [Google Scholar] [CrossRef]
- Mai, P.L.; Best, A.F.; Peters, J.A.; DeCastro, R.M.; Khincha, P.P.; Loud, J.T.; Bremer, R.C.; Rosenberg, P.S.; Savage, S.A. Risks of first and subsequent cancers among TP53 mutationcarriers in the NCI LFS cohort. Cancer 2016, 122, 3673–3681. [Google Scholar] [CrossRef]
- Manoukian, S.; Peissel, B.; Frigerio, S.; Lecis, D.; Bartkova, J.; Roversi, G.; Radice, P.; Bartek, J.; Delia, D. Two new CHEK2 germline variants detected in breast cancer/sarcoma families negative for BRCA1, BRCA2, and TP53 gene mutations. Breast Cancer Res. Treat. 2011, 130, 207–215. [Google Scholar] [CrossRef]
- Calvete, O.; Garcia-Pavia, P.; Domínguez, F.; Bougeard, G.; Kunze, K.; Braeuninger, A.; Teule, A.; Lasa, A.; Ramon y Cajal, T.; Llort, G.; et al. The wide spectrum of POT1 gene variants correlates with multiple cancer types. Eur. J. Hum. Genet. 2017, 25, 1278–1281. [Google Scholar] [CrossRef]
- Vietri, M.T.; Molinari, A.M.; Caliendo, G.; De Paola, M.L.; D’Elia, G.; Gambardella, A.L.; Petronella, P.; Cioffi, M. Double heterozygosity in the BRCA1 and BRCA2 genes in Italian family. Clin. Chem. Lab. Med. 2013, 51, 2319–2324. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.T.; Caliendo, G.; Schiano, C.; Casamassimi, A.; Molinari, A.M.; Napoli, C.; Cioffi, M. Analysis of PALB2 in a cohort of Italian breast cancer patients: Identification of a novel PALB2 truncating mutation. Fam. Cancer 2015, 14, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.T.; Caliendo, G.; Casamassimi, A.; Cioffi, M.; De Paola, M.L.; Napoli, C.; Molinari, A.M. A novel PALB2 truncating mutation in an Italian family with male breast cancer. Oncol. Rep. 2015, 33, 1243–1247. [Google Scholar] [CrossRef]
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V.; et al. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.T.; Miccoli, S.; Turchetti, D.; Bondavalli, D.; Viel, A.; Quaia, M.; Giacomini, E.; Gismondi, V.; Sanchez-Mete, L.; Stigliano, V.; et al. Type and frequency of MUTYH variants in Italian patients with suspected MAP: A retrospective multicenter study. J. Hum. Genet. 2017, 62, 309–315. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, V.G.; Minoprio, A.; Torreri, P.; Marinoni, I.; Bossa, C.; Petrucci, T.C.; Albertini, A.M.; Ranzani, G.N.; Bignami, M.; Mazzei, F. Functional analysis of MUTYH mutated proteins associated with familial adenomatous polyposis. DNA Repair 2010, 9, 700–707. [Google Scholar] [CrossRef]
- Kurian, A.W.; Hare, E.E.; Mills, M.A.; Kingham, K.E.; McPherson, L.; Whittemore, A.S.; McGuire, V.; Ladabaum, U.; Kobayashi, Y.; Lincoln, S.E.; et al. Clinical Evaluation of a Multiple-Gene Sequencing Panel for Hereditary Cancer Risk Assessment. J. Clin. Oncol. 2014, 32, 2001–2009. [Google Scholar] [CrossRef]
- Velázquez, C.; Lastra, E.; Avila Cobos, F.; Abella, L.; de la Cruz, V.; Hernando, B.A.; Hernández, L.; Martínez, N.; Infante, M.; Durán, M. A comprehensive custom panel evaluation for routine hereditary cancer testing: Improving the yield of germline mutation detection. J. Transl. Med. 2020, 18, 232. [Google Scholar] [CrossRef]
- Zografos, E.; Andrikopoulou, A.; Papatheodoridi, A.M.; Kaparelou, M.; Bletsa, G.; Liontos, M.; Dimopoulos, M.A.; Zagouri, F. Multi-Gene Mutation Profiling by Targeted Next-Generation Sequencing in Premenopausal Breast Cancer. Genes 2022, 13, 1362. [Google Scholar] [CrossRef]
- Easton, D.F.; Pharoah, P.D.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-panel sequencing and the prediction of breast-cancer risk. N. Engl. J. Med. 2015, 372, 2243–2257. [Google Scholar] [CrossRef]
- Kamps, R.; Brandão, R.D.; Bosch, B.J.; Paulussen, A.D.; Xanthoulea, S.; Blok, M.J.; Romano, A. Next-Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification. Int. J. Mol. Sci. 2017, 18, 308. [Google Scholar] [CrossRef] [PubMed]
- Kanth, P.; Grimmett, J.; Champine, M.; Burt, R.; Samadder, N.J. Hereditary colorectal polyposis and cancer syndromes: A primer on diagnosis and management. Am. J. Gastroenterol. 2017, 112, 1509–1525. [Google Scholar] [CrossRef] [PubMed]
- Daly, M.B.; Pal, T.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Goggins, M.; Hutton, M.L.; et al. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2021, 19, 77–102. [Google Scholar] [CrossRef] [PubMed]
- Pinto, C.; Biffoni, M.; Popoli, P.; Marchetti, A.; Marchetti, P.; Martini, N.; Normanno, N. Molecular tests and target therapies in oncology: Recommendations from the Italian workshop. Future Oncol. 2021, 17, 3529–3539. [Google Scholar] [CrossRef]
- Montisci, A.; Vietri, M.T.; Palmieri, V.; Sala, S.; Donatelli, F.; Napoli, C. Cardiac Toxicity Associated with Cancer Immuno-therapy and Biological Drugs. Cancers 2021, 13, 4797. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Elia, G.; Caliendo, G.; Passariello, L.; Albanese, L.; Makker, J.; Molinari, A.M.; Vietri, M.T. Hereditary Cancer Syndrome in a Family with Double Mutation in BRIP1 and MUTYH Genes. Genes 2023, 14, 428. https://doi.org/10.3390/genes14020428
D’Elia G, Caliendo G, Passariello L, Albanese L, Makker J, Molinari AM, Vietri MT. Hereditary Cancer Syndrome in a Family with Double Mutation in BRIP1 and MUTYH Genes. Genes. 2023; 14(2):428. https://doi.org/10.3390/genes14020428
Chicago/Turabian StyleD’Elia, Giovanna, Gemma Caliendo, Luana Passariello, Luisa Albanese, Jasmine Makker, Anna Maria Molinari, and Maria Teresa Vietri. 2023. "Hereditary Cancer Syndrome in a Family with Double Mutation in BRIP1 and MUTYH Genes" Genes 14, no. 2: 428. https://doi.org/10.3390/genes14020428
APA StyleD’Elia, G., Caliendo, G., Passariello, L., Albanese, L., Makker, J., Molinari, A. M., & Vietri, M. T. (2023). Hereditary Cancer Syndrome in a Family with Double Mutation in BRIP1 and MUTYH Genes. Genes, 14(2), 428. https://doi.org/10.3390/genes14020428