
Global Distribution of Founder Variants Associated with Non-Syndromic Hearing Impairment
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Search Strategy
2.2. Selection Criteria and Data Extraction
2.3. Quality Assessment
2.4. Pathogenicity and Clinical Significance
3. Results
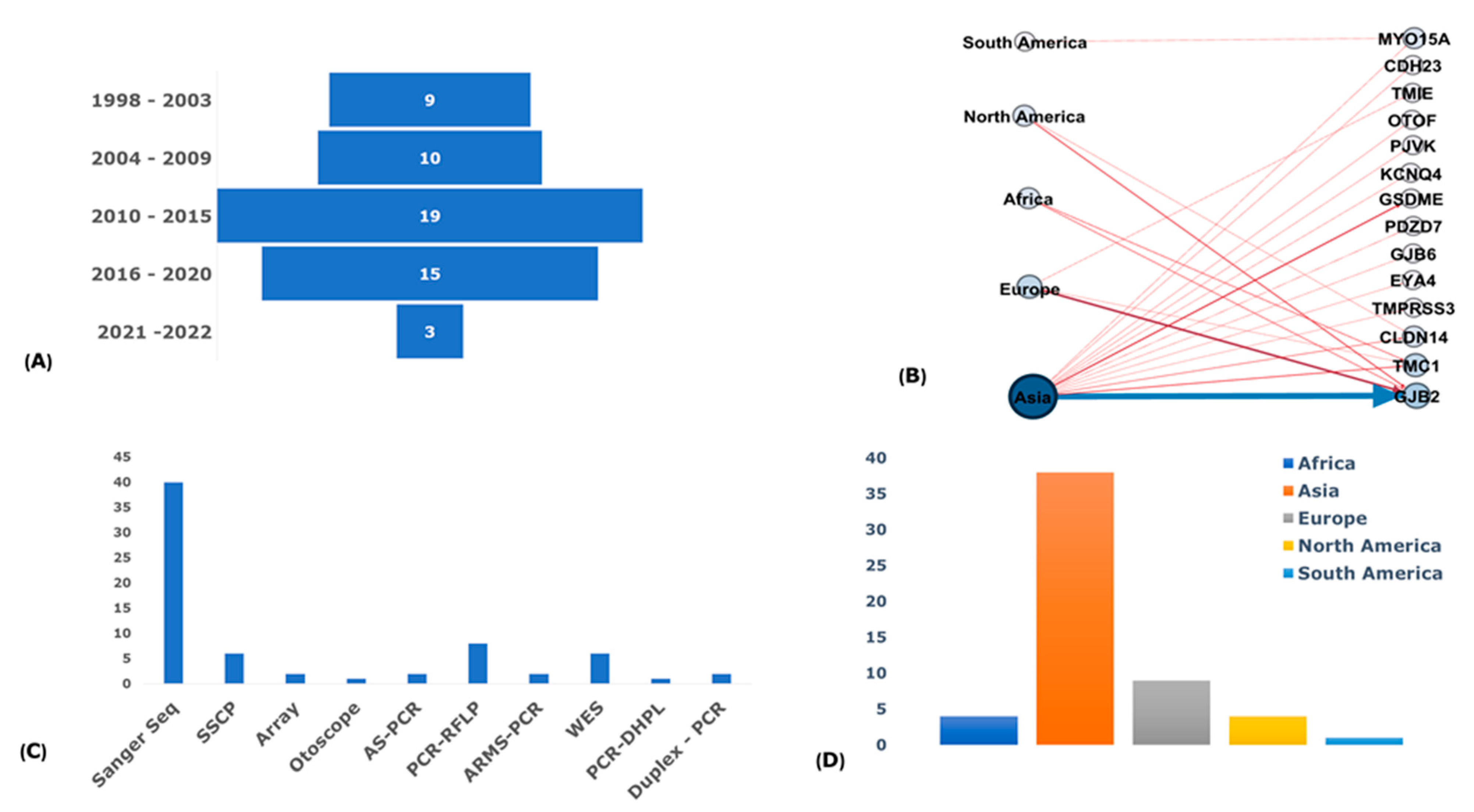
3.1. Publication Search Outcome

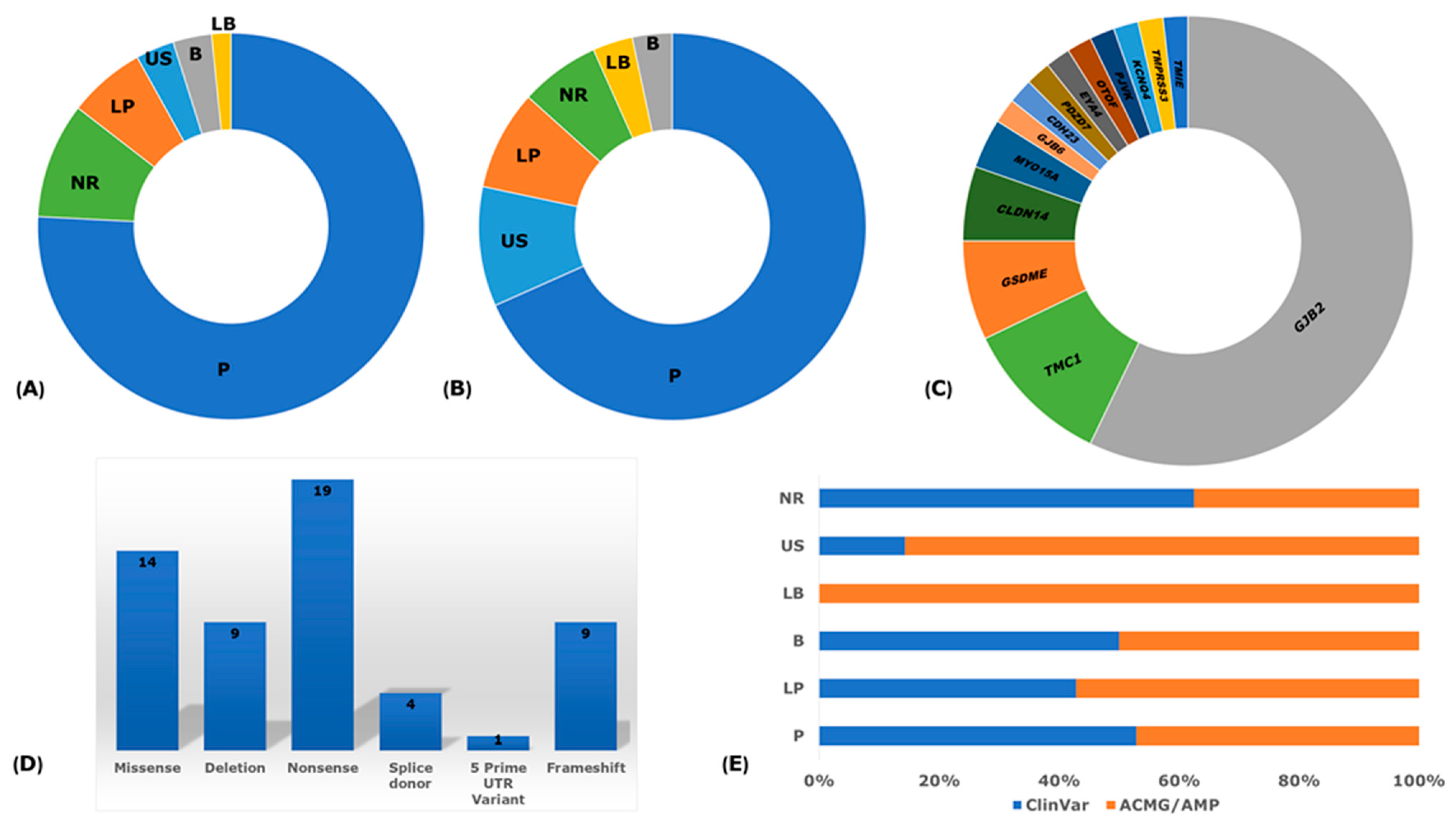
3.2. Biased and Quality Assessment
3.3. Publications and Participants Descriptions
3.4. Non-Syndromic Hearing Impairment Phenotyping
3.5. Genotyping Methods
3.6. Haplotype and Marker Analysis
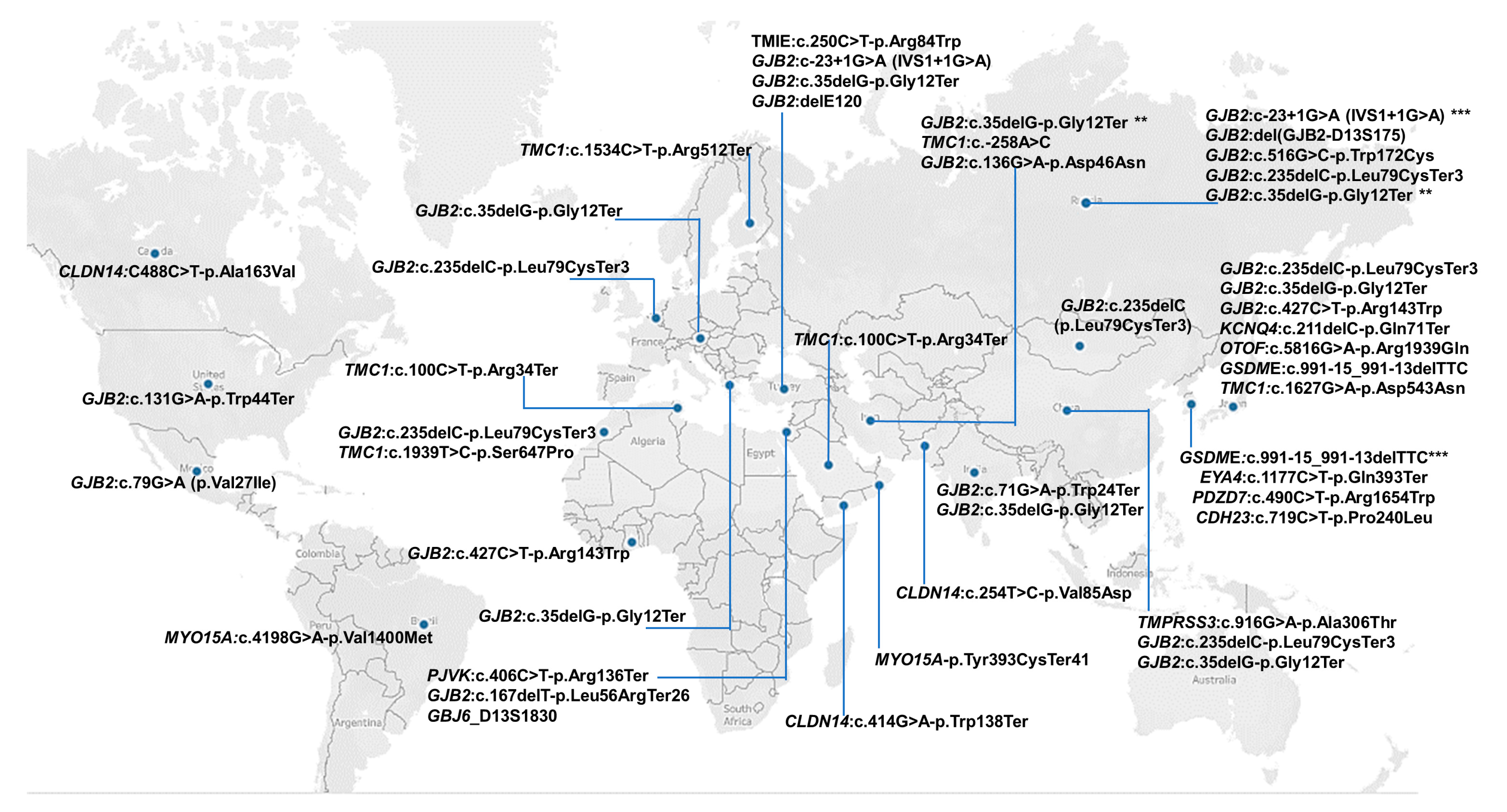
3.7. Regional Distribution of Founder Variants
3.8. Reported Founder Mutation Genes
3.9. Pathological Mechanisms of Implicated Genes
4. Discussion
4.1. GJB2 Founder Variants
4.2. Other Genes (Non-GJB2) Founder Variants
4.3. General Observations on Founder Variants
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bliznetz, E.A.; Lalayants, M.R.; Markova, T.G.; Balanovsky, O.P.; Balanovska, E.V.; Skhalyakho, R.A.; Pocheshkhova, E.A.; Nikitina, N.V.; Voronin, S.V.; Kudryashova, E.K. Update of the GJB2/DFNB1 Mutation Spectrum in Russia: A Founder Ingush Mutation Del (GJB2-D13S175) Is the Most Frequent among Other Large Deletions. J. Hum. Genet. 2017, 62, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Mikstiene, V.; Jakaitiene, A.; Byckova, J.; Gradauskiene, E.; Preiksaitiene, E.; Burnyte, B.; Tumiene, B.; Matuleviciene, A.; Ambrozaityte, L.; Uktveryte, I. The High Frequency of GJB2 Gene Mutation c. 313_326del14 Suggests Its Possible Origin in Ancestors of Lithuanian Population. BMC Genet. 2016, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Borck, G.; Rainshtein, L.; Hellman-Aharony, S.; Volk, A.E.; Friedrich, K.; Taub, E.; Magal, N.; Kanaan, M.; Kubisch, C.; Shohat, M. High Frequency of Autosomal-Recessive DFNB59 Hearing Loss in an Isolated Arab Population in Israel. Clin. Genet. 2012, 82, 271–276. [Google Scholar] [CrossRef]
- Kenneson, A.; Braun, K.V.N.; Boyle, C. GJB2 (Connexin 26) Variants and Nonsyndromic Sensorineural Hearing Loss: A HuGE Review. Genet. Med. 2002, 4, 258–274. [Google Scholar] [CrossRef] [PubMed]
- Hilgert, N.; Smith, R.J.; Van Camp, G. Forty-Six Genes Causing Nonsyndromic Hearing Impairment: Which Ones Should Be Analyzed in DNA Diagnostics? Mutat. Res. Rev. Mutat. Res. 2009, 681, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Denoyelle, F.; Weil, D.; Maw, M.A.; Wilcox, S.A.; Lench, N.J.; Allen-Powell, D.R.; Osborn, A.H.; Dahl, H.-H.M.; Middleton, A.; Houseman, M.J. Prelingual Deafness: High Prevalence of a 30delG Mutation in the Connexin 26 Gene. Hum. Mol. Genet. 1997, 6, 2173–2177. [Google Scholar] [CrossRef]
- Frei, K.; Szuhai, K.; Lucas, T.; Weipoltshammer, K.; Schöfer, C.; Ramsebner, R.; Baumgartner, W.-D.; Raap, A.K.; Bittner, R.; Wachtler, F.J. Connexin 26 Mutations in Cases of Sensorineural Deafness in Eastern Austria. Eur. J. Hum. Genet. 2002, 10, 427–432. [Google Scholar] [CrossRef]
- Gürtler, N.; Kim, Y.; Mhatre, A.; Müller, R.; Probst, R.; Lalwani, A.K. GJB2 Mutations in the Swiss Hearing Impaired. Ear Hear. 2003, 24, 440–447. [Google Scholar] [CrossRef]
- Bouwer, S.; Angelicheva, D.; Chandler, D.; Seeman, P.; Tournev, I.; Kalaydjieva, L. Carrier Rates of the Ancestral Indian W24X Mutation in GJB2 in the General Gypsy Population and Individual Subisolates. Genet. Test. 2007, 11, 455–458. [Google Scholar] [CrossRef]
- Wonkam, E.T.; Chimusa, E.; Noubiap, J.J.; Adadey, S.M.; Fokouo, J.V.F.; Wonkam, A. GJB2 and GJB6 Mutations in Hereditary Recessive Non-Syndromic Hearing Impairment in Cameroon. Genes 2019, 10, 844. [Google Scholar] [CrossRef]
- Brobby, G.W.; Müller-Myhsok, B.; Horstmann, R.D. Connexin 26 R143W Mutation Associated with Recessive Nonsyndromic Sensorineural Deafness in Africa. N. Engl. J. Med. 1998, 338, 548–550. [Google Scholar] [CrossRef] [PubMed]
- Hamelmann, C.; Amedofu, G.K.; Albrecht, K.; Muntau, B.; Gelhaus, A.; Brobby, G.W.; Horstmann, R.D. Pattern of Connexin 26 (GJB2) Mutations Causing Sensorineural Hearing Impairment in Ghana. Hum. Mutat. 2001, 18, 84–85. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Bajaj, Y.; Sirimanna, T.; Albert, D.M.; Qadir, P.; Jenkins, L.; Bitner-Glindzicz, M. Spectrum of GJB2 Mutations Causing Deafness in the British Bangladeshi Population. Clin. Otolaryngol. 2008, 33, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Coyle, B.; Reardon, W.; Herbrick, J.-A.; Tsui, L.-C.; Gausden, E.; Lee, J.; Coffey, R.; Grueters, A.; Grossman, A.; Phelps, P.D. Molecular Analysis of the PDS Gene in Pendred Syndrome (Sensorineural Hearing Loss and Goitre). Hum. Mol. Genet. 1998, 7, 1105–1112. [Google Scholar] [CrossRef]
- Tsukamoto, K.; Suzuki, H.; Harada, D.; Namba, A.; Abe, S.; Usami, S. Distribution and Frequencies of PDS (SLC26A4) Mutations in Pendred Syndrome and Nonsyndromic Hearing Loss Associated with Enlarged Vestibular Aqueduct: A Unique Spectrum of Mutations in Japanese. Eur. J. Hum. Genet. 2003, 11, 916–922. [Google Scholar] [CrossRef]
- Park, H.J.; Shaukat, S.; Liu, X.Z.; Hahn, S.H.; Naz, S.; Ghosh, M.; Kim, H.N.; Moon, S.K.; Abe, S.; Tukamoto, K. Origins and Frequencies of SLC26A4 (PDS) Mutations in East and South Asians: Global Implications for the Epidemiology of Deafness. J. Med. Genet. 2003, 40, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C.; Yeh, T.-H.; Chen, P.-J.; Hsu, C.-J. Prevalent SLC26A4 Mutations in Patients with Enlarged Vestibular Aqueduct and/or Mondini Dysplasia: A Unique Spectrum of Mutations in Taiwan, Including a Frequent Founder Mutation. Laryngoscope 2005, 115, 1060–1064. [Google Scholar] [CrossRef]
- Dai, P.; Li, Q.; Huang, D.; Yuan, Y.; Kang, D.; Miller, D.T.; Shao, H.; Zhu, Q.; He, J.; Yu, F. SLC26A4 c. 919-2A> G Varies among Chinese Ethnic Groups as a Cause of Hearing Loss. Genet. Med. 2008, 10, 586–592. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Group, T.P. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef]
- Sohani, Z.N.; Meyre, D.; de Souza, R.J.; Joseph, P.G.; Gandhi, M.; Dennis, B.B.; Norman, G.; Anand, S.S. Assessing the Quality of Published Genetic Association Studies in Meta-Analyses: The Quality of Genetic Studies (Q-Genie) Tool. BMC Genet. 2015, 16, 1–8. [Google Scholar] [CrossRef]
- Hoy, D.; Brooks, P.; Woolf, A.; Blyth, F.; March, L.; Bain, C.; Baker, P.; Smith, E.; Buchbinder, R. Assessing Risk of Bias in Prevalence Studies: Modification of an Existing Tool and Evidence of Interrater Agreement. J. Clin. Epidemiol. 2012, 65, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Shinagawa, J.; Moteki, H.; Nishio, S.; Noguchi, Y.; Usami, S. Haplotype Analysis of GJB2 Mutations: Founder Effect or Mutational Hot Spot? Genes 2020, 11, 250. [Google Scholar] [CrossRef]
- Morell, R.J.; Kim, H.J.; Hood, L.J.; Goforth, L.; Friderici, K.; Fisher, R.; Van Camp, G.; Berlin, C.I.; Oddoux, C.; Ostrer, H. Mutations in the Connexin 26 Gene (GJB2) among Ashkenazi Jews with Nonsyndromic Recessive Deafness. N. Engl. J. Med. 1998, 339, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Laer, L.V.; Coucke, P.; Mueller, R.F.; Caethoven, G.; Flothmann, K.; Prasad, S.D.; Chamberlin, G.P.; Houseman, M.; Taylor, G.R.; de Heyning, C.M.V.; et al. A Common Founder for the 35delG GJB2 Gene Mutation in Connexin 26 Hearing Impairment. J. Med. Genet. 2001, 38, 515–518. [Google Scholar] [CrossRef]
- Janecke, A.R.; Hirst-Stadlmann, A.; Günther, B.; Utermann, B.; Müller, T.; Löffler, J.; Utermann, G.; Nekahm-Heis, D. Progressive Hearing Loss, and Recurrent Sudden Sensorineural Hearing Loss Associated with GJB2 Mutations–Phenotypic Spectrum and Frequencies of GJB2 Mutations in Austria. Hum. Genet. 2002, 111, 145–153. [Google Scholar] [CrossRef]
- Tekin, M.; Duman, T.; Boğoçlu, G.; İncesulu, A.; Çomak, E.; Ilhan, I.; Akar, N. Spectrum of GJB2 Mutations in Turkey Comprises Both Caucasian and Oriental Variants: Roles of Parental Consanguinity and Assortative Mating. Hum. Mutat. 2003, 21, 552–553. [Google Scholar] [CrossRef]
- Qi, L.; Pu, D.; De-Liang, H.; Jin, Z.; Guo-Jian, W.; Qing-Wen, Z.; Xin, L.; Dong-Yi, H. Prevalence of the GJB2 Mutations in Deafness Patients of Different Ethnic Origins in Xinjiang. J. Otol. 2007, 2, 23–29. [Google Scholar] [CrossRef]
- Abidi, O.; Boulouiz, R.; Nahili, H.; Imken, L.; Rouba, H.; Chafik, A.; Barakat, A. The Analysis of Three Markers Flanking GJB2 Gene Suggests a Single Origin of the Most Common 35delG Mutation in the Moroccan Population. Biochem. Biophys. Res. Commun. 2008, 377, 971–974. [Google Scholar] [CrossRef]
- Kokotas, H.; Van Laer, L.; Grigoriadou, M.; Iliadou, V.; Economides, J.; Pomoni, S.; Pampanos, A.; Eleftheriades, N.; Ferekidou, E.; Korres, S. Strong Linkage Disequilibrium for the Frequent GJB2 35delG Mutation in the Greek Population. Am. J. Med. Genet. Part A 2008, 146, 2879–2884. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, G.; Bhalla, S.; Khullar, M.; Panda, N.K. High Frequency of Heterozygosity in GJB2 Mutations among Patients with Non-Syndromic Hearing Loss. J. Laryngol. Otol. 2009, 123, 273. [Google Scholar] [CrossRef] [PubMed]
- Dzhemileva, L.U.; Barashkov, N.A.; Posukh, O.L.; Khusainova, R.I.; Akhmetova, V.L.; Kutuev, I.A.; Gilyazova, I.R.; Tadinova, V.N.; Fedorova, S.A.; Khidiyatova, I.M. Carrier Frequency of GJB2 Gene Mutations c. 35delG, c. 235delC and c. 167delT among the Populations of Eurasia. J. Hum. Genet. 2010, 55, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Norouzi, V.; Azizi, H.; Fattahi, Z.; Esteghamat, F.; Bazazzadegan, N.; Nishimura, C.; Nikzat, N.; Jalalvand, K.; Kahrizi, K.; Smith, R.J. Did the GJB2 35delG Mutation Originate in Iran? Am. J. Med. Genet. Part A 2011, 155, 2453–2458. [Google Scholar] [CrossRef] [PubMed]
- Zytsar, M.V.; Barashkov, N.A.; Bady-Khoo, M.S.; Shubina-Olejnik, O.A.; Danilenko, N.G.; Bondar, A.A.; Morozov, I.V.; Solovyev, A.V.; Danilchenko, V.Y.; Maximov, V.N.; et al. Updated Carrier Rates for c.35delG (GJB2) Associated with Hearing Loss in Russia and Common c.35delG Haplotypes in Siberia. BMC Med. Genet. 2018, 19, 138. [Google Scholar] [CrossRef]
- Naddafnia, H.; Noormohammadi, Z.; Irani, S.; Salahshoorifar, I. Frequency of GJB2 Mutations, GJB6-D13S1830 and GJB6-D13S1854 Deletions among Patients with Non-Syndromic Hearing Loss from the Central Region of Iran. Mol. Genet. Genom. Med. 2019, 7, 1–8. [Google Scholar]
- Yan, D.; Park, H.-J.; Ouyang, X.M.; Pandya, A.; Doi, K.; Erdenetungalag, R.; Du, L.L.; Matsushiro, N.; Nance, W.E.; Griffith, A.J. Evidence of a Founder Effect for the 235delC Mutation of GJB2 (Connexin 26) in East Asians. Hum. Genet. 2003, 114, 44–50. [Google Scholar] [CrossRef]
- Ohtsuka, A.; Yuge, I.; Kimura, S.; Namba, A.; Abe, S.; Van Laer, L.; Van Camp, G.; Usami, S. GJB2 Deafness Gene Shows a Specific Spectrum of Mutations in Japan, Including a Frequent Founder Mutation. Hum. Genet. 2003, 112, 329–333. [Google Scholar] [CrossRef]
- Posukh, O.; Pallares-Ruiz, N.; Tadinova, V.; Osipova, L.; Claustres, M.; Roux, A.-F. First Molecular Screening of Deafness in the Altai Republic Population. BMC Med. Genet. 2005, 6, 12. [Google Scholar] [CrossRef]
- Zytsar, M.V.; Bady-Khoo, M.S.; Danilchenko, V.Y.; Maslova, E.A.; Barashkov, N.A.; Morozov, I.V.; Bondar, A.A.; Posukh, O.L. High Rates of Three Common GJB2 Mutations c.516G>C, c.−23+1G>A, c.235delC in Deaf Patients from Southern Siberia Are Due to the Founder Effect. Genes 2020, 11, 833. [Google Scholar] [CrossRef]
- del Castillo, I.; Moreno-Pelayo, M.A.; del Castillo, F.J.; Brownstein, Z.; Marlin, S.; Adina, Q.; Cockburn, D.J.; Pandya, A.; Siemering, K.R.; Chamberlin, G.P.; et al. Prevalence and Evolutionary Origins of the Del(GJB6-D13S1830) Mutation in the DFNB1 Locus in Hearing-Impaired Subjects: A Multicenter Study. Am. J. Hum. Genet. 2003, 73, 1452–1458. [Google Scholar] [CrossRef]
- del Castillo, F.J.; Rodríguez-Ballesteros, M.; Álvarez, A.; Hutchin, T.; Leonardi, E.; de Oliveira, C.A.; Azaiez, H.; Brownstein, Z.; Avenarius, M.R.; Marlin, S.; et al. A Novel Deletion Involving the Connexin-30 Gene, Del (GJB6-D13s1854), Found in Trans with Mutations in the GJB2 Gene (Connexin-26) in Subjects with DFNB1 Non-Syndromic Hearing Impairment. J. Med. Genet. 2005, 42, 588–594. [Google Scholar] [CrossRef]
- Tekin, M.; Boğoclu, G.; Arican, S.T.; Orman, M.N.; Tastan, H.; Elsayed, S.; Akar, N. Evidence for Single Origins of 35delG and DelE120 Mutations in the GJB2 Gene in Anatolia. Clin. Genet. 2005, 67, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.Y.; Rasool, T.J. High Frequency of Connexin26 (GJB2) Mutations Associated with Nonsyndromic Hearing Loss in the Population of Kerala, India. Int. J. Pediatr. Otorhinolaryngol. 2009, 73, 437–443. [Google Scholar] [CrossRef]
- García-Sánchez, G.; Alfaro-Rodríguez, A.; Poblano, A. Evidence for Central Asian Origin of the p. Val27Ile Variant in the GJB2 Gene. Int. J. Med. Genet. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Carranza, C.; Menendez, I.; Herrera, M.; Castellanos, P.; Amado, C.; Maldonado, F.; Rosales, L.; Escobar, N.; Guerra, M.; Alvarez, D. A Mayan Founder Mutation Is a Common Cause of Deafness in Guatemala. Clin. Genet. 2016, 89, 461–465. [Google Scholar] [CrossRef]
- Palombo, F.; Al-Wardy, N.; Ruscone, G.A.G.; Oppo, M.; Kindi, M.N.A.; Angius, A.; Al Lamki, K.; Girotto, G.; Giangregorio, T.; Benelli, M.; et al. A Novel Founder MYO15A Frameshift Duplication Is the Major Cause of Genetic Hearing Loss in Oman. J. Hum. Genet. 2017, 62, 259–264. [Google Scholar] [CrossRef]
- Manzoli, G.N.; Bademci, G.; Acosta, A.X.; Félix, T.M.; Cengiz, F.B.; Foster, J.; Da Silva, D.S.D.; Menendez, I.; Sanchez-Pena, I.; Tekin, D. Targeted Resequencing of Deafness Genes Reveals a Founder MYO15A Variant in Northeastern Brazil. Ann. Hum. Genet. 2016, 80, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Sirmaci, A.; Akcayoz-Duman, D.; Tekin, M. The c. IVS1+ 1G> A Mutation Inthe GJB2 Gene Is Prevalent and Large Deletions Involving the GJB6 Gene Are Not Present in the Turkish Population. J. Genet. 2006, 85, 213–216. [Google Scholar] [CrossRef]
- Davoudi-Dehaghani, E.; Zeinali, S.; Mahdieh, N.; Shirkavand, A.; Bagherian, H.; Tabatabaiefar, M.A. A Transversion Mutation in Non-Coding Exon 3 of the TMC1 Gene in Two Ethnically Related Iranian Deaf Families from Different Geographical Regions; Evidence for Founder Effect. Int. J. Pediatr. Otorhinolaryngol. 2013, 77, 821–826. [Google Scholar] [CrossRef]
- Bazazzadegan, N.; Sheffield, A.M.; Sobhani, M.; Kahrizi, K.; Meyer, N.C.; Van Camp, G.; Hilgert, N.; Abedini, S.S.; Habibi, F.; Daneshi, A. Two Iranian Families with a Novel Mutation in GJB2 Causing Autosomal Dominant Nonsyndromic Hearing Loss. Am. J. Med. Genet. Part A 2011, 155, 1202–1211. [Google Scholar] [CrossRef]
- Ramzan, K.; Al-Owain, M.; Al-Numair, N.S.; Afzal, S.; Al-Ageel, S.; Al-Amer, S.; Al-Baik, L.; Al-Otaibi, G.F.; Hashem, A.; Al-Mashharawi, E. Identification of TMC1 as a Relatively Common Cause for Nonsyndromic Hearing Loss in the Saudi Population. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2020, 183, 172–180. [Google Scholar] [CrossRef]
- Saïd, M.B.; Hmani-Aifa, M.; Amar, I.; Baig, S.M.; Mustapha, M.; Delmaghani, S.; Tlili, A.; Ghorbel, A.; Ayadi, H.; Van Camp, G. High Frequency of the p. R34X Mutation in the TMC1 Gene Associated with Nonsyndromic Hearing Loss Is Due to Founder Effects. Genet. Test. Mol. Biomark. 2010, 14, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Huang, S.-S.; Yuan, Y.-Y.; Xu, J.-C.; Gu, P.; Bai, D.; Kang, D.-Y.; Han, M.-Y.; Wang, G.-J.; Zhang, M.-G. Identification of TMPRSS3 as a Significant Contributor to Autosomal Recessive Hearing Loss in the Chinese Population. Neural Plast. 2017, 2017, 3192090. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, T.; Mutai, H.; Kunishima, S.; Namba, K.; Morimoto, N.; Shinjo, Y.; Arimoto, Y.; Kataoka, Y.; Shintani, T.; Morita, N. A Prevalent Founder Mutation and Genotype–Phenotype Correlations of OTOF in Japanese Patients with Auditory Neuropathy. Clin. Genet. 2012, 82, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, A.R.; Kim, N.K.; Kim, M.Y.; Jeon, E.-H.; Kim, B.J.; Han, Y.E.; Chang, M.Y.; Park, W.-Y.; Choi, B.Y. Strong Founder Effect of p. P240L in CDH23 in Koreans and Its Significant Contribution to Severe-to-Profound Nonsyndromic Hearing Loss in a Korean Pediatric Population. J. Transl. Med. 2015, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bashir, Z.-H.; Latief, N.; Belyantseva, I.A.; Iqbal, F.; Amer Riazuddin, S.; Khan, S.N.; Friedman, T.B.; Riazuddin, S.; Riazuddin, S. Phenotypic Variability of CLDN14 Mutations Causing DFNB29 Hearing Loss in the Pakistani Population. J. Hum. Genet. 2013, 58, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Pater, J.A.; Benteau, T.; Griffin, A.; Penney, C.; Stanton, S.G.; Predham, S.; Kielley, B.; Squires, J.; Zhou, J.; Li, Q. A Common Variant in CLDN14 Causes Precipitous, Prelingual Sensorineural Hearing Loss in Multiple Families Due to Founder Effect. Hum. Genet. 2017, 136, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, W.K.E.; Mahfood, M.; Al Mutery, A.; Abdallah, S.H.; Tlili, A. A Novel Nonsense Mutation (c. 414G> A; p. Trp138*) in CLDN14 Causes Hearing Loss in Yemeni Families: A Case Report. Front. Genet. 2019, 10, 1087. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Han, J.H.; Kim, B.J.; Oh, S.H.; Lee, S.; Oh, D.-Y.; Choi, B.Y. Identification of a Potential Founder Effect of a Novel PDZD7 Variant Involved in Moderate-to-Severe Sensorineural Hearing Loss in Koreans. Int. J. Mol. Sci. 2019, 20, 4174. [Google Scholar] [CrossRef]
- Barashkov, N.A.; Dzhemileva, L.U.; Fedorova, S.A.; Teryutin, F.M.; Posukh, O.L.; Fedotova, E.E.; Lobov, S.L.; Khusnutdinova, E.K. Autosomal Recessive Deafness 1A (DFNB1A) in Yakut Population Isolate in Eastern Siberia: Extensive Accumulation of the Splice Site Mutation IVS1+ 1G> A in GJB2 Gene as a Result of Founder Effect. J. Hum. Genet. 2011, 56, 631–639. [Google Scholar] [CrossRef]
- Bonyadi, M.; Fotouhi, N.; Esmaeili, M. Prevalence of IVS1+1G>A Mutation among Iranian Azeri Turkish Patients with Autosomal Recessive Non-Syndromic Hearing Loss (ARNSHL). Int. J. Pediatr. Otorhinolaryngol. 2011, 75, 1612–1615. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, A.V.; Kushniarevich, A.; Bliznetz, E.; Bady-Khoo, M.; Lalayants, M.R.; Markova, T.G.; Minárik, G.; Metspalu, E.; Pshennikova, V.G.; Teryutin, F.M. A Common Founder Effect of the Splice Site Variant c.−23+ 1G> A in GJB2 Gene Causing Autosomal Recessive Deafness 1A (DFNB1A) in Eurasia. Hum. Genet. 2022, 141, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Nishio, S.; Iwasa, Y.; Yano, T.; Kumakawa, K.; Abe, S.; Ishikawa, K.; Kojima, H.; Namba, A.; Oshikawa, C. Comprehensive Genetic Screening of KCNQ4 in a Large Autosomal Dominant Nonsyndromic Hearing Loss Cohort: Genotype-Phenotype Correlations and a Founder Mutation. PLOS ONE 2013, 8, e63231. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Meng, X.; Zhang, S.; Zhao, G.; Hu, L.; Kong, X. A 3-Nucleotide Deletion in the Polypyrimidine Tract of Intron 7 of the DFNA5 Gene Causes Nonsyndromic Hearing Impairment in a Chinese Family. Genomics 2003, 82, 575–579. [Google Scholar] [CrossRef]
- Park, H.-J.; Cho, H.-J.; Baek, J.-I.; Ben-Yosef, T.; Kwon, T.-J.; Griffith, A.J.; Kim, U.-K. Evidence for a Founder Mutation Causing DFNA5 Hearing Loss in East Asians. J. Hum. Genet. 2010, 55, 59–62. [Google Scholar] [CrossRef]
- Nishio, A.; Noguchi, Y.; Sato, T.; Naruse, T.K.; Kimura, A.; Takagi, A.; Kitamura, K. A DFNA5 Mutation Identified in Japanese Families with Autosomal Dominant Hereditary Hearing Loss. Ann. Hum. Genet. 2014, 78, 83–91. [Google Scholar] [CrossRef]
- Booth, K.T.; Azaiez, H.; Smith, R.J. DFNA5 (GSDME) c. 991-15_991-13delTTC: Founder Mutation or Mutational Hotspot? Int. J. Mol. Sci. 2020, 21, 3951. [Google Scholar] [CrossRef]
- Brownstein, Z.; Friedman, L.M.; Shahin, H.; Oron-Karni, V.; Kol, N.; Rayyan, A.A.; Parzefall, T.; Lev, D.; Shalev, S.; Frydman, M. Targeted Genomic Capture and Massively Parallel Sequencing to Identify Genes for Hereditary Hearing Loss in Middle Eastern Families. Genome Biol. 2011, 12, 1–11. [Google Scholar] [CrossRef]
- Kraatari-Tiri, M.; Haanpää, M.K.; Willberg, T.; Pohjola, P.; Keski-Filppula, R.; Kuismin, O.; Moilanen, J.S.; Häkli, S.; Rahikkala, E. Clinical and Genetic Characteristics of Finnish Patients with Autosomal Recessive and Dominant Non-Syndromic Hearing Loss Due to Pathogenic TMC1 Variants. J. Clin. Med. 2022, 11, 1837. [Google Scholar] [CrossRef]
- Nishio, S.; Usami, S. Prevalence and Clinical Features of Autosomal Dominant and Recessive TMC1-Associated Hearing Loss. Hum. Genet. 2022, 141, 929–937. [Google Scholar] [CrossRef]
- Slatkin, M.; Rannala, B. Estimating Allele Age. Annu. Rev. Genom. Hum. Genet. 2000, 1, 225–249. [Google Scholar] [CrossRef] [PubMed]
- Reeve, J.P.; Rannala, B. DMLE+: Bayesian Linkage Disequilibrium Gene Mapping. Bioinformatics 2002, 18, 894–895. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xia, X.; Ke, X.; Ouyang, X.; Du, L.; Liu, Y.; Angeli, S.; Telischi, F.F.; Nance, W.E.; Balkany, T.; et al. The Prevalence of Connexin 26 (GJB2) Mutations in the Chinese Population. Hum. Genet. 2002, 111, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, P.; Rabionet, R.; Barbujani, G.; Melchionda, S.; Petersen, M.; Brøndum-Nielsen, K.; Metspalu, A.; Oitmaa, E.; Pisano, M.; Fortina, P. High Carrier Frequency of the 35delG Deafness Mutation in European Populations. Eur. J. Hum. Genet. 2000, 8, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Tekin, M.; Xia, X.-J.; Erdenetungalag, R.; Cengiz, F.B.; White, T.W.; Radnaabazar, J.; Dangaasuren, B.; Tastan, H.; Nance, W.E.; Pandya, A. GJB2 Mutations in Mongolia: Complex Alleles, Low Frequency, and Reduced Fitness of the Deaf. Ann. Hum. Genet. 2010, 74, 155–164. [Google Scholar] [CrossRef]
- Aboagye, E.T.; Adadey, S.M.; Esoh, K.; Jonas, M.; de Kock, C.; Amenga-Etego, L.; Awandare, G.A.; Wonkam, A. Age Estimate of GJB2-p.(Arg143Trp) Founder Variant in Hearing Impairment in Ghana, Suggests Multiple Independent Origins across Populations. Biology 2022, 11, 476. [Google Scholar] [CrossRef]
- Wingard, J.C.; Zhao, H.-B. Cellular and Deafness Mechanisms Underlying Connexin Mutation-Induced Hearing Loss–a Common Hereditary Deafness. Front. Cell. Neurosci. 2015, 9, 202. [Google Scholar] [CrossRef]
- Wang, H.-L.; Chang, W.-T.; Li, A.H.; Yeh, T.-H.; Wu, C.-Y.; Chen, M.-S.; Huang, P.-C. Functional Analysis of Connexin-26 Mutants Associated with Hereditary Recessive Deafness. J. Neurochem. 2003, 84, 735–742. [Google Scholar] [CrossRef]
- Zhang, J.; Scherer, S.S.; Yum, S.W. Dominant Cx26 Mutants Associated with Hearing Loss Have Dominant-Negative Effects on Wild Type Cx26. Mol. Cell. Neurosci. 2011, 47, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Bicego, M.; Beltramello, M.; Melchionda, S.; Carella, M.; Piazza, V.; Zelante, L.; Bukauskas, F.F.; Arslan, E.; Cama, E.; Pantano, S. Pathogenetic Role of the Deafness-Related M34T Mutation of Cx26. Hum. Mol. Genet. 2006, 15, 2569–2587. [Google Scholar] [CrossRef]
- Liu, Y.-P.; Zhao, H.-B. Cellular Characterization of Connexin26 and Connnexin30 Expression in the Cochlear Lateral Wall. Cell Tissue Res. 2008, 333, 395–403. [Google Scholar] [CrossRef]
- Cohen-Salmon, M.; Ott, T.; Michel, V.; Hardelin, J.-P.; Perfettini, I.; Eybalin, M.; Wu, T.; Marcus, D.C.; Wangemann, P.; Willecke, K. Targeted Ablation of Connexin26 in the Inner Ear Epithelial Gap Junction Network Causes Hearing Impairment and Cell Death. Curr. Biol. 2002, 12, 1106–1111. [Google Scholar] [CrossRef]
- Meyer, C.G.; Amedofu, G.K.; Brandner, J.M.; Pohland, D.; Timmann, C.; Horstmann, R.D. Selection for Deafness? Nat. Med. 2002, 8, 1332–1333. [Google Scholar] [CrossRef] [PubMed]
- Snoeckx, R.L.; Huygen, P.L.; Feldmann, D.; Marlin, S.; Denoyelle, F.; Waligora, J.; Mueller-Malesinska, M.; Pollak, A.; PLoSki, R.; Murgia, A. GJB2 Mutations and Degree of Hearing Loss: A Multicenter Study. Am. J. Hum. Genet. 2005, 77, 945–957. [Google Scholar] [CrossRef]
- Del Castillo, F.J.; Del Castillo, I. DFNB1 Non-Syndromic Hearing Impairment: Diversity of Mutations and Associated Phenotypes. Front. Mol. Neurosci. 2017, 10, 428. [Google Scholar] [CrossRef]
- Esoh, K.; Wonkam, A. Evolutionary History of Sickle-Cell Mutation: Implications for Global Genetic Medicine. Hum. Mol. Genet. 2021, 30, R119–R128. [Google Scholar] [CrossRef] [PubMed]
- Schrijver, I. Hereditary Non-Syndromic Sensorineural Hearing Loss: Transforming Silence to Sound. J. Mol. Diagn. 2004, 6, 275–284. [Google Scholar] [CrossRef]
- Abe, S.; Usami, S.; Shinkawa, H.; Kelley, P.M.; Kimberling, W.J. Prevalent Connexin 26 Gene (GJB2) Mutations in Japanese. J. Med. Genet. 2000, 37, 41–43. [Google Scholar] [CrossRef] [PubMed]
- Essamak, B.F. Non-Syndromic Autosomal Recessive Deafness in Gaza Strip: A Study on Selected Connexin 26 Gene Mutations. Int. J. Genet. Genomics. 2014, 2, 92–96. [Google Scholar] [CrossRef]
- Yuan, Y.; Huang, D.; Yu, F.; Zhu, X.; Kang, D.; Yuan, H.; Han, D.; Dai, P. A de Novo GJB2 (Connexin 26) Mutation, R75W, in a Chinese Pedigree with Hearing Loss and Palmoplantar Keratoderma. Am. J. Med. Genet. Part A 2009, 149, 689–692. [Google Scholar] [CrossRef]
- Shimkin, D.B. Siberian Ethnography: Historical Sketch and Evaluation. Arct. Anthropol. 1990, 27, 36–51. [Google Scholar]
- Mannai-ool, M.K. Tuvan People. The Origin and Formation of the Ethnos; Nauka Publishing: Novosibirsk, Russia, 2004; pp. 99–166. [Google Scholar]
- Kenna, M.A.; Feldman, H.A.; Neault, M.W.; Frangulov, A.; Wu, B.-L.; Fligor, B.; Rehm, H.L. Audiologic Phenotype and Progression in GJB2 (Connexin 26) Hearing Loss. Arch. Otolaryngol. Head Neck Surg. 2010, 136, 81–87. [Google Scholar] [CrossRef]
- Wonkam, A.; Adadey, S.M.; Schrauwen, I.; Aboagye, E.T.; Wonkam-Tingang, E.; Esoh, K.; Popel, K.; Manyisa, N.; Jonas, M.; deKock, C.; et al. Exome Sequencing of Families from Ghana Reveals Known and Candidate Hearing Impairment Genes. Commun. Biol. 2022, 5, 1–16. [Google Scholar] [CrossRef]
- Tsukada, K.; Nishio, S.; Hattori, M.; Usami, S. Ethnic-Specific Spectrum of GJB2 and SLC26A4 Mutations: Their Origin and a Literature Review. Ann. Otol. Rhinol. Laryngol. 2015, 124, 61S–76S. [Google Scholar] [CrossRef]
- Dia, Y.; Adadey, S.M.; Diop, J.P.D.; Aboagye, E.T.; Ba, S.A.; De Kock, C.; Ly, C.A.T.; Oluwale, O.G.; Sène, A.R.G.; Sarr, P.D.; et al. GJB2 Is a Major Cause of Non-Syndromic Hearing Impairment in Senegal. Biology 2022, 11, 795. [Google Scholar] [CrossRef] [PubMed]
- Adeyemo, A.; Faridi, R.; Chattaraj, P.; Yousaf, R.; Tona, R.; Okorie, S.; Bharadwaj, T.; Nouel-Saied, L.M.; Acharya, A.; Schrauwen, I. Genomic Analysis of Childhood Hearing Loss in the Yoruba Population of Nigeria. Eur. J. Hum. Genet. 2022, 30, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Wonkam, A.; Manyisa, N.; Bope, C.D.; Dandara, C.; Chimusa, E.R. Whole Exome Sequencing Reveals Pathogenic Variants in MYO3A, MYO15A and COL9A3 and Differential Frequencies in Ancestral Alleles in Hearing Impairment Genes among Individuals from Cameroon. Hum. Mol. Genet. 2020, 29, 3729–3743. [Google Scholar] [CrossRef]
- Yalcouyé, A.; Traoré, O.; Taméga, A.; Maïga, A.B.; Kané, F.; Oluwole, O.G.; Guinto, C.O.; Kéita, M.; Timbo, S.K.; DeKock, C.; et al. Etiologies of Childhood Hearing Impairment in Schools for the Deaf in Mali. Front. Pediatr. 2021, 9, 1367. [Google Scholar] [CrossRef]
- Sırmacı, A.; Öztürkmen-Akay, H.; Erbek, S.; Incesulu, A.; Duman, D.; Taşır-Yılmaz, S.; Özdağ, H.; Tekin, M. A Founder TMIE Mutation Is a Frequent Cause of Hearing Loss in Southeastern Anatolia. Clin. Genet. 2009, 75, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Park, S.M.; Chang, S.O.; Chung, T.; Lee, K.Y.; Kim, A.R.; Park, J.H.; Kim, V.; Park, W.-Y.; Oh, S.-H. A Novel Mutation of TMPRSS3 Related to Milder Auditory Phenotype in Korean Postlingual Deafness: A Possible Future Implication for a Personalized Auditory Rehabilitation. J. Mol. Med. 2014, 92, 651–663. [Google Scholar] [CrossRef]
- Basel-Vanagaite, L.; Taub, E.; Halpern, G.J.; Drasinover, V.; Magal, N.; Davidov, B.; Zlotogora, J.; Shohat, M. Genetic Screening for Autosomal Recessive Nonsyndromic Mental Retardation in an Isolated Population in Israel. Eur. J. Hum. Genet. 2007, 15, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Cavalli-Sforza, L.L.; Feldman, M.W. The Application of Molecular Genetic Approaches to the Study of Human Evolution. Nat. Genet. 2003, 33, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Searle, C.; Mavrogiannis, L.A.; Bennett, C.P.; Charlton, R.S. The Common TMC1 Mutation c. 100C> T (p. Arg34X) Is Not a Significant Cause of Deafness in British Asians. Genet. Test. Mol. Biomark. 2012, 16, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Nance, W.E.; Liu, X.-Z.; Pandya, A. Relation between Choice of Partner and High Frequency of Connexin-26 Deafness. Lancet 2000, 356, 500–501. [Google Scholar] [CrossRef]
- Arcos-Burgos, M.; Muenke, M. Genetics of Population Isolates. Clin. Genet. 2002, 61, 233–247. [Google Scholar] [CrossRef]
- Chong, J.X.; Ouwenga, R.; Anderson, R.L.; Waggoner, D.J.; Ober, C. A Population-Based Study of Autosomal-Recessive Disease-Causing Mutations in a Founder Population. Am. J. Hum. Genet. 2012, 91, 608–620. [Google Scholar] [CrossRef]
- Evans, J.A. Old Meets New: Identifying Founder Mutations in Genetic Disease. CMAJ 2015, 187, 93–94. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ExAC Freq | GnomAD Exomes Freq | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference SNP | Transcript | Locus | Founder Variant | Coding Impact | ACMG/AMP | ClinVar | Ref. Allele | Alt. Allele | Ref. Allele | Alt.Allele | Ref |
| rs80338948 | NM_004004.6 | DFNB1 | GJB2: c.427C>T-p.Arg143Trp | Missense | P (PP5, PM1, PM2, PP3, PM2) | P | 0.999835 | 0.000165 | 0.999884 | 0.000116 | [11,23] |
| rs80338942 | NM_004004.6 | DFNB1 | GJB2: c.167delT-p.Leu56ArgTer26 | Frameshift | P (PSV1, PP5) | P | 0.999316 | 0.000684 | 0.999415 | 0.000585 | [24] |
| rs80338939 | NM_004004.6 | DFNB1 | GJB2: c.35delG-p.Gy12Ter | Nonsense | P (PSV1, PM2, PP5) | P | 0.993960 | 0.006040 | 0.993443 | 0.006557 | [25,26,27,28,29,30,31,32,33,34,35] |
| rs80338943 | NM_004004.6 | DFNB1 | GJB2: c.235delC-p.Leu79CysTer3 | Frameshift | P (PSVB1 PS3, PP5, PM2) | P | 0.999637 | 0.000363 | 0.999531 | 0.000469 | [25,28,36,37,38,39] |
| NA | NA | DFNB1 | GJB6: ∆(GJB6-D13S1830) | Deletion | NR | NR | NR | NR | NR | NR | [40,41] |
| NA | NA | DFNB1 | GJB2: delE120 | Deletion | NR | NR | NR | NR | NR | NR | [42] |
| rs104894396 | NM_004004.6 | DFNB1 | GJB2: c.71G>A-p.Trp24Ter | Nonsense | P (PSV1, PP55, PM2) | P | 0.999423 | 0.000577 | 0.999416 | 0.000584 | [43] |
| rs2274084 | NM_004004.6 | DFNB1 | GJB2: c.79G>A-p.Val27Ile | Missense | B (BA1, PP6, BP4, PM1) | B | 0.954619 | 0.045381 | 0.945994 | 0.054006 | [44] |
| rs104894413 | NM_004004.6 | DFNB1 | GJB2: c.131G>A-p.Trp44Ter | Nonsense | P (PS3, PM1, PP3, PM5, PM2) | P | 0.999992 | 0.000008 | 0.999976 | 0.000024 | [45] |
| NA | NA | DFNB1 | GJB2: del(GJB2-D13S175) | Deletion | NR | P | NR | NR | NR | NR | [1] |
| rs1302739538 | NM_004004.6 | DFNB1 | GJB2: c.516G>C-p.Trp172Cys | Missense | P (PP5, PM1, PM2, PP3, PM2) | P/LP | NMR | NR | NR | NR | [39] |
| rs1567620939 | NM_016239.4 | DFNB3 | MYO15A: c.1171_1177dupGCCATCT -p.Tyr393CysTer41 | Frameshift | P (PVS1, PP5, PM2) | P | NR | NR | NR | NR | [46] |
| rs749136456 | NM_016239.4 | DFNB3 | MYO15A: c.4198G>A-p.Val1400Met | Missense | P (PP5, PP3, PM2,BP1) | LP | NR | NR | NR | NR | [47] |
| rs28942097 | NM_147196.3 | DFNB6 | TMIE: c.250C>T-p.Arg84Trp | Missense | P/LP (PP5, PM3, PP3, PM2) | P/LP | 0.999983 | 0.000017 | 0.999976 | 0.000024 | [48] |
| rs937270834 | NM_138691.3 | DFNB7/11 | TMC1: c.-258A>C | 5 Prime UTR Variant | LB (BP4, PM2) | NR | NR | NR | NR | NR | [49] |
| rs121908073 | NM_138691.3 | DFNB7/11 | TMC1: c.100C > T-p.Arg34Ter | Nonsense | P (PVS1, PP5, PM2) | P | 0.999948 | 0.000052 | 0.999944 | 0.000056 | [50,51,52] |
| rs181949335 | NM_032404.2 | DFNB8 | TMPRSS3: c.916G>A-p.Ala306Thr | Missense | P (PP5, PM1, PM5, PP3) | P/LP | 0.999827 | 0.000173 | 0.999855 | 0.000145 | [53] |
| rs80356605 | NM_194248.3 | DFNB9 | OTOF: c.5816G>A-p.Arg1939Gln | Missense | US (PP5, PM2, PP3, BP1) | P | NR | NR | 0.999957 | 0.000043 | [54] |
| rs121908354 | NM_022124.6 | DFNB12 | CDH23: c.C719T-p.Pro240Leu | Missense | LP (PP5, PM1, PM2, BP4) | P | 0.999909 | 0.000091 | 0.999960 | 0.000040 | [55] |
| rs74315437 | NM_012130.4 | DFNB29 | CLDN14: c.254T>A-p.Val85Asp | Missense | US (PM5, PM2, PP3) | P | 0.999992 | 0.000008 | 1.000000 | 0.000000 | [56] |
| rs143797113 | NM_012130.4 | DFNB29 | CLDN14: c.488C>T-p.Ala163Val | Missense | US (PM2, PP5) | US, LP | 0.999744 | 0.000256 | 0.999708 | 0.000292 | [57] |
| rs142846225 | NM_012130.4 | DFNB29 | CLDN14: c.414G>A-p.Trp138Ter | Nonsense | P (PVS1, PP5, PM2) | NR | 0.999992 | 0.000008 | 0.999988 | 0.000012 | [58] |
| rs200664140 | NM_001195263.2 | DFNB57 | PDZD7: c.490C > T-p.Arg164Trp | Missense | US (PM2, PP5, BP1) | US | NR | NR | NR | NR | [59] |
| rs367688416 | NM_001042702.5 | DFNB59 | PJVK: c.406C>T-p.Arg136Ter | Nonsense | P (PSV1, PP5, PM2) | P | 0.999982 | 0.000018 | 0.999984 | 0.000016 | [3] |
| rs80338940 | NM_004004.6 | DFNBA1 | GJB2: c.-23+1G>A | Splice donor | P (PSV1, PP5. PM2) | P | NR | NR | 0.999829 | 0.000171 | [39,60,61,62] |
| rs80358272 | NM_004700.4 | DFNA2 | KCNQ4: c.211delC-p.Gln71SerTer68 | Nonsense | P (PSV1, PM2, PP5) | P | NR | NR | NR | NR | [63] |
| rs1064797088 | NM_004004.6 | DFNA3A | GJB2: c.136G>A-p.Asp46Asn | Missense | P (PM1, PM5, PP3, PP5, PM2) | P | NR | NR | NR | NR | [50] |
| rs727505273 | NM_001127453.2 | DFNA5 | GSDME: c.991-15_991-13delTTC | Deletion | LP (PP5, PM2, BP4) | P | NR | NR | NR | NR | [64,65,66,67] |
| rs757172581 | NM_004100.5 | DFNA10 | EYA4: c.1177C>T-p.Gln393Ter | Nonsense | P (PVS1, PM2, PP5) | NR | 0.999992 | 0.000008 | 0.999996 | 0.000004 | [55] |
| rs138527651 | NM_138691.3 | DFNA36 | TMC1: c.1939T>C-p.Ser647Pro | Missense | US (PP5, PM2, PP3) | P | 0.999992 | 0.000008 | 0.999976 | 0.000024 | [68] |
| rs200171616 | NM_138691.3 | DFNA36 | TMC1: c.1534C>T-p.Arg512Ter | Nonsense | P (PVS1, PM2, PP3, PP5) | P | 0.999802 | 0.000198 | 0.999670 | 0.000330 | [69] |
| NA | NM_138691.3 | DFNA36 | TMC1: c.1627G>A-p.Asp543Asn | Missense | US (PM2, PP3) | US, LP | NR | NR | NR | NR | [70] |
| Gene | Category | Expected SNVs | Observed SNVs | Constraint Metrics |
|---|---|---|---|---|
| GJB2 | Synonymous Missense pLoF | 61.1 137.1 6.5 | 64 161 17 | Z = −0.29 o/e = 1.05 (0.86–1.29) Z = −0.72 o/e = 1.17 (1.03–1.34) pLI = 0 o/e = 2.62 (1.39–1.98) |
| GJB6 | Synonymous Missense pLoF | 64 149.6 9.3 | 69 134 10 | Z = 0.99, o/e = 1.08 (0.89–1.32) Z = 0.45, o/e = 0.9 (0.78–1.03) pLI = 0, o/e = 1.07 (0.66–1.74) |
| GSDME | Synonymous Missense pLoF | 116.1 265.4 21.6 | 128 304 16 | Z = −0.87, o/e = 1.1 (0.95–1.28) Z = −0.84, o/e = 1.15 (1.04–1.26) pLI = 0, o/e = 0.74 (0.5–1.13) |
| CDH23 | Synonymous Missense pLoF | 301.3 715.2 47.1 | 290 662 18 | Z = 0.51, o/e = 0.96 (0.87–1.06) Z = 0.71, o/e = 0.93 (0.87–0.99) pLI = 0, o/e = 0.38 (0.26–0.57) |
| CLDN14 | Synonymous Missense pLoF | 77 154.9 6.2 | 81 148 6 | Z = −0.35, o/e = 1.05 (0.88–1.26) Z = 0.2, o/e = 0.96 (0.83–1.09) pLI = 0, o/e = 0.97 (0.52–1.75) |
| MYO15A | Synonymous Missense pLoF | 886.4 2057.4 169.1 | 826 1975 116 | Z = 1.6, o/e = 0.93 (0.88–0.99) Z = 0.65, o/e = 0.96 (0.92–1) pLI = 0, o/e = 0.69 (0.59–0.8) |
| PDZD7 | Synonymous Missense pLoF | 138.9 330 22.5 | 134 139 17 | Z = 0.32, o/e = 0.96 (0.84–1.11) Z = −0.37, o/e = 1.06 (0.97–1.16) pLI = 0, o/e = 0.76 (0.52–1.13) |
| EYA4 | Synonymous Missense pLoF | 122.5 333.4 7.7 | 276 103 10 | Z = 1.38, o/e = 0.84 (0.72–0.99) Z = 1.12, o/e = 0.83 (0.75–0.92) pLI = 0.05, o/e = 0.27 (0.16–0.45) |
| PJVK | Synonymous Missense pLoF | 64.9 186.1 19.1 | 57 181 14 | Z = 0.77, o/e = 0.88 (0.71–1.09) Z = 0.13, o/e = 0.97 (0.86–1.1) pLI = 0, o/e = 0.73 (0.48–1.15) |
| TMC1 | Synonymous Missense pLoF | 142.1 398.2 47.6 | 146 350 35 | Z = −0.26;, o/e = 1.03 (0.9–1.18) Z = 0.86, o/e = 0.88 (0.81–0.96) pLI = 0, o/e = 0.74 (0.56–0.97) |
| KCNQ4 | Synonymous Missense pLoF | 179 412.2 32.1 | 156 288 7 | Z = 1.35, o/e = 0.87 (0.76–0.99) Z = 2.17, o/e = 0.7 (0.63–0.77) pLI = 0.47, o/e = 0.22 (0.12–0.41) |
| TMPRSS3 | Synonymous Missense pLoF | 101.9 253.3 24.3 | 94 212 17 | Z = 0.62, o/e = 0.92 (0.78–1.09) Z = 0.92, o/e = 0.84 (0.75–0.94) pLI = 0, o/e = 0.7 (0.48–1.05) |
| TMIE | Synonymous Missense pLoF | 31.2 75.3 5.7 | 25 74 6 | Z = 0.87, o/e = 0.8 (0.58–1.12) Z = 0.05, o/e = 0.98 (0.81–1.19) pLI = 0, o/e = 1.05 (0.57–1.81) |
| OTOF | Synonymous Missense pLoF | 508.8 1223.6 105.2 | 550 1252 76 | Z = −1.44, o/e = 1.08 (1.01–1.16) Z = −0.29, o/e = 1.02 (0.98–1.07) pLI = 0, o/e = 0.72 (0.6–0.88) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aboagye, E.T.; Adadey, S.M.; Wonkam-Tingang, E.; Amenga-Etego, L.; Awandare, G.A.; Wonkam, A. Global Distribution of Founder Variants Associated with Non-Syndromic Hearing Impairment. Genes 2023, 14, 399. https://doi.org/10.3390/genes14020399
Aboagye ET, Adadey SM, Wonkam-Tingang E, Amenga-Etego L, Awandare GA, Wonkam A. Global Distribution of Founder Variants Associated with Non-Syndromic Hearing Impairment. Genes. 2023; 14(2):399. https://doi.org/10.3390/genes14020399
Chicago/Turabian StyleAboagye, Elvis Twumasi, Samuel Mawuli Adadey, Edmond Wonkam-Tingang, Lucas Amenga-Etego, Gordon A. Awandare, and Ambroise Wonkam. 2023. "Global Distribution of Founder Variants Associated with Non-Syndromic Hearing Impairment" Genes 14, no. 2: 399. https://doi.org/10.3390/genes14020399
APA StyleAboagye, E. T., Adadey, S. M., Wonkam-Tingang, E., Amenga-Etego, L., Awandare, G. A., & Wonkam, A. (2023). Global Distribution of Founder Variants Associated with Non-Syndromic Hearing Impairment. Genes, 14(2), 399. https://doi.org/10.3390/genes14020399