
Expanding Genotype/Phenotype Correlation in 2p11.2-p12 Microdeletion Syndrome


Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Molecular Genetic Analysis
2.3. Literature Search
3. Results
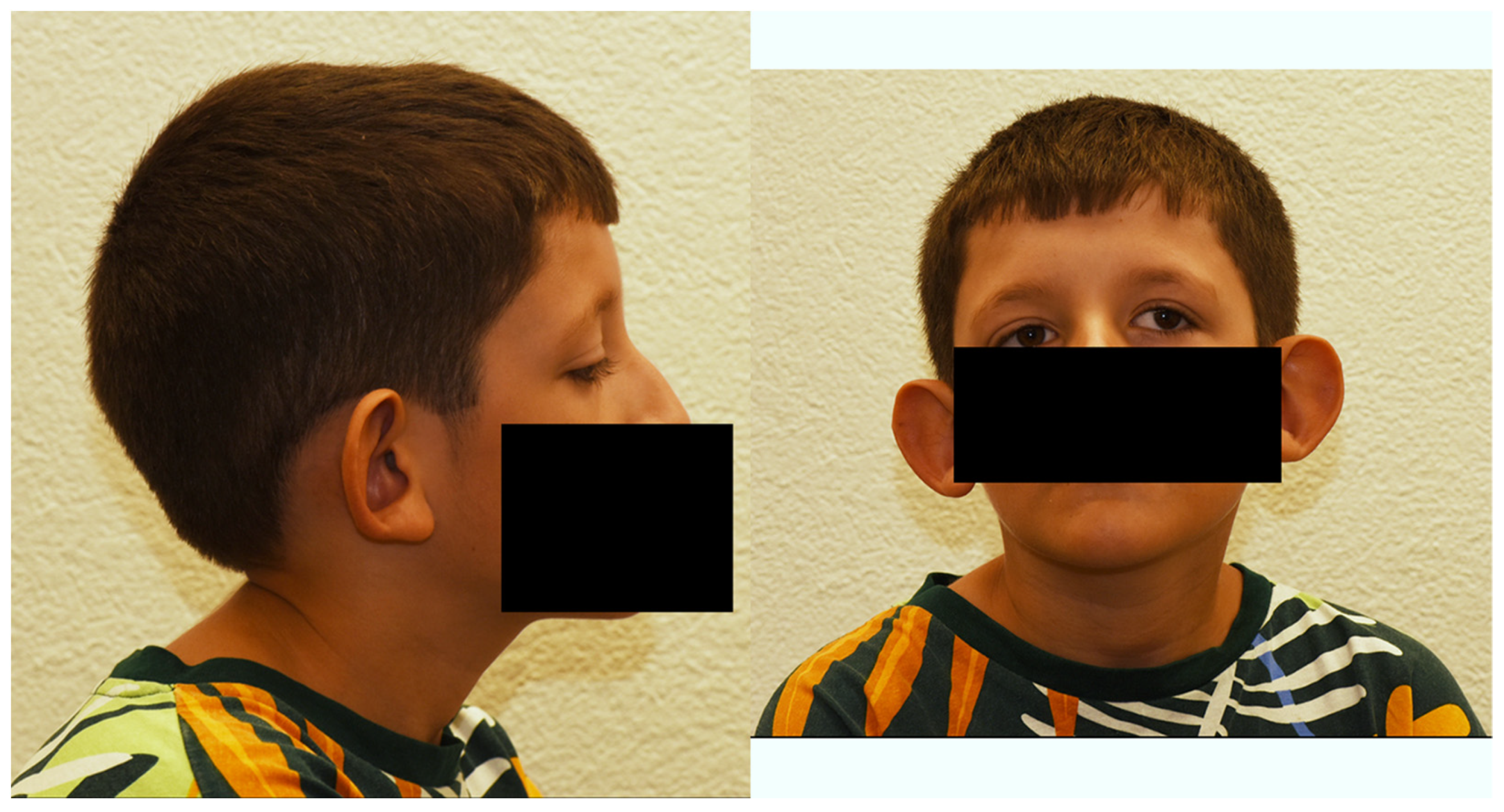
3.1. Clinical Case Report
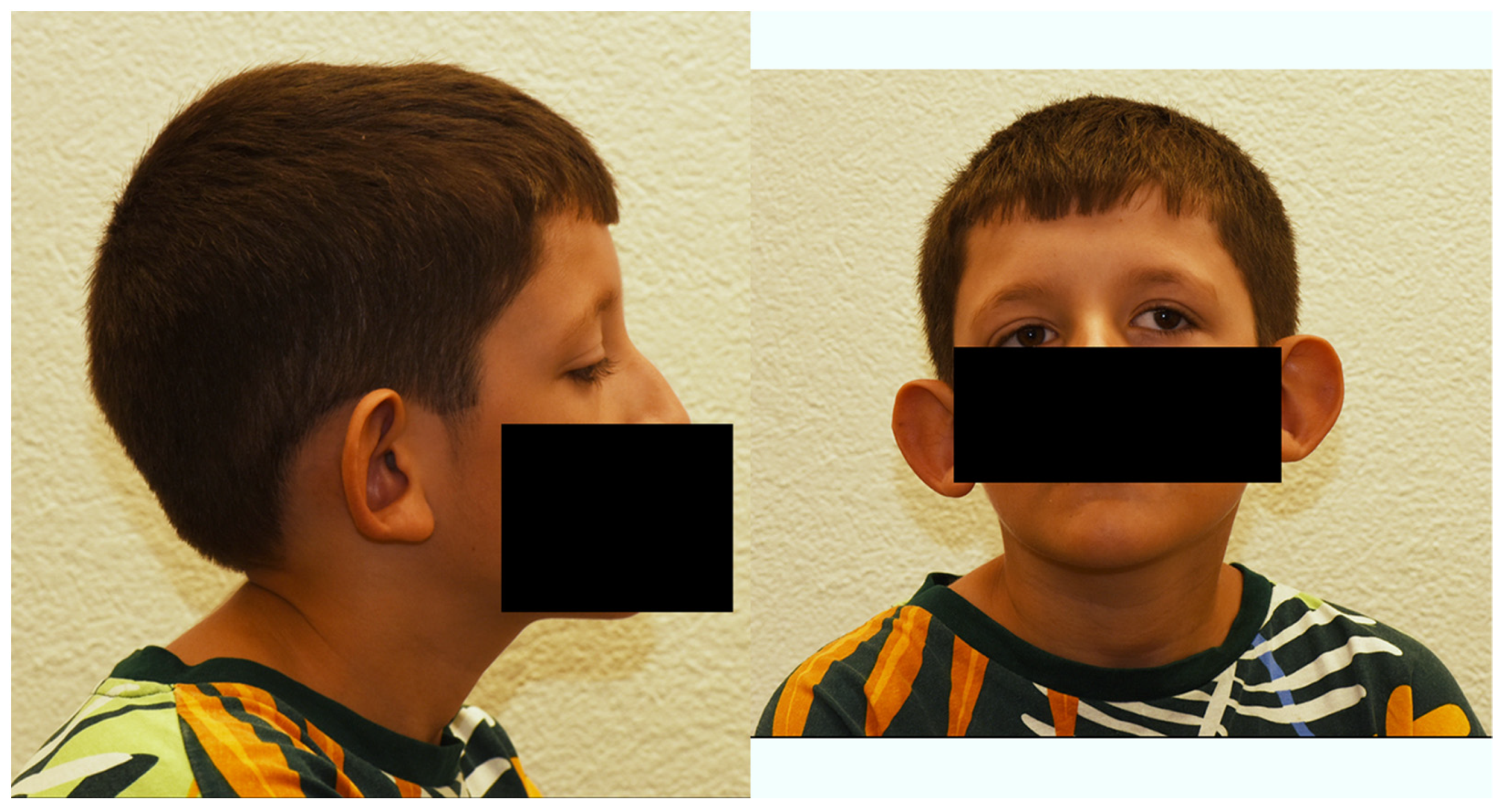
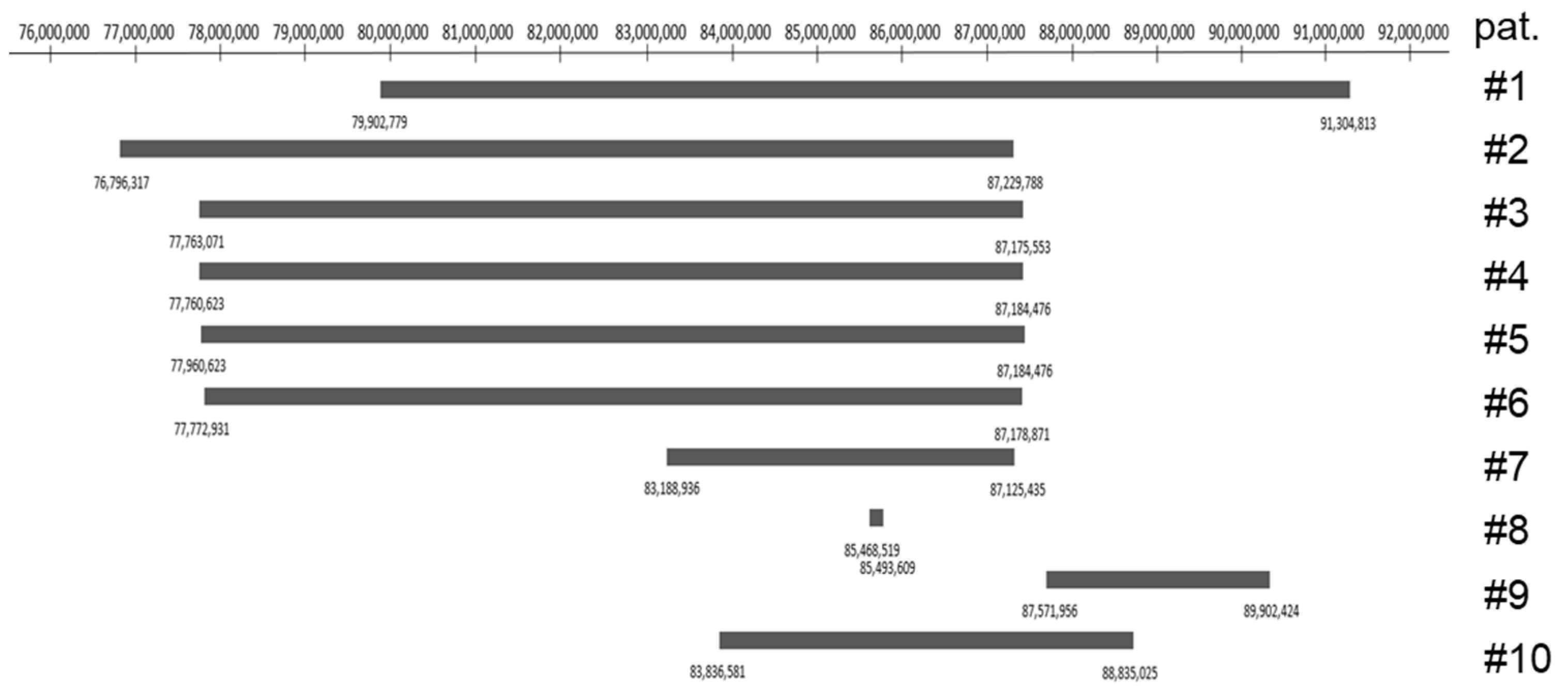
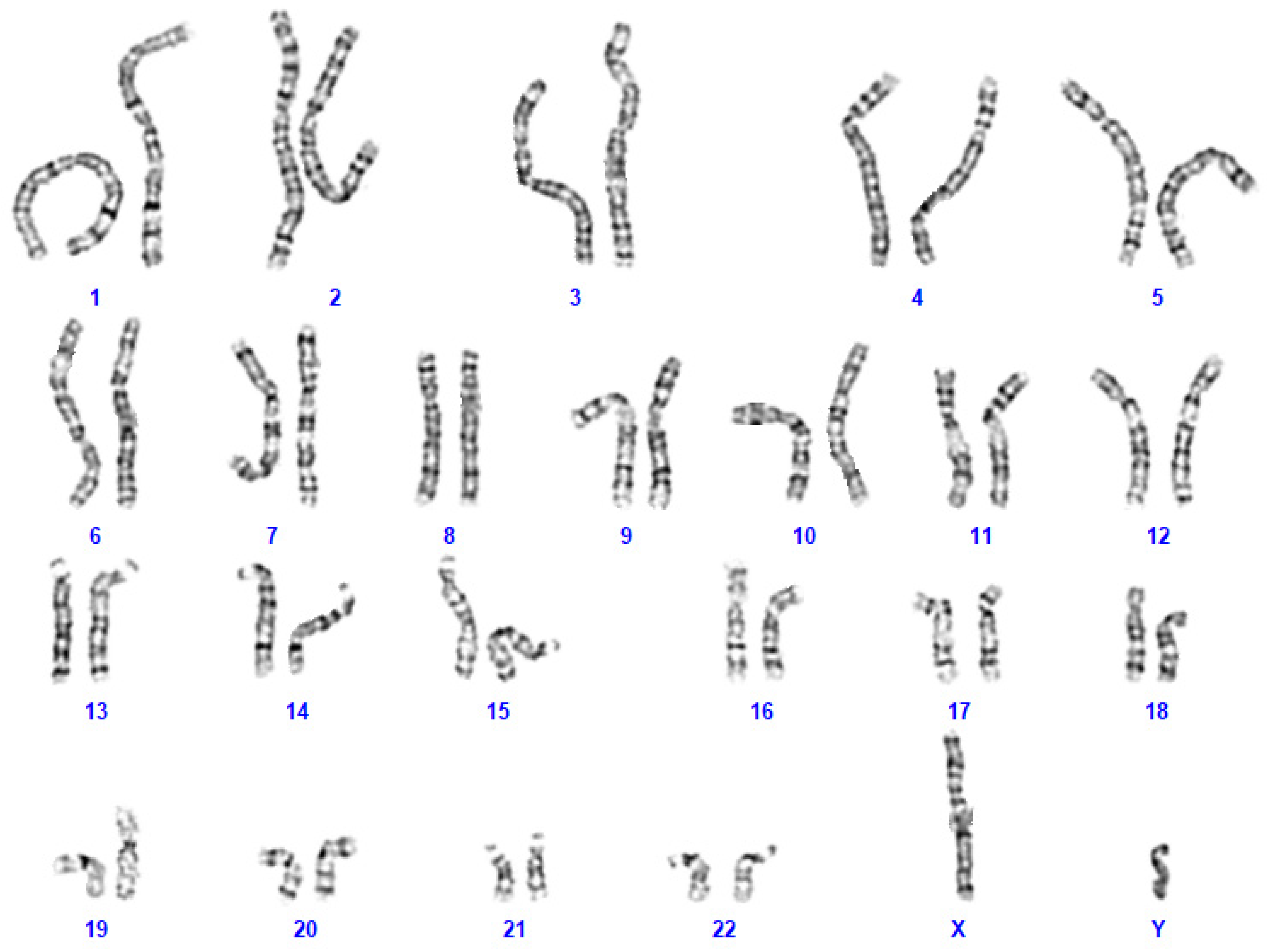
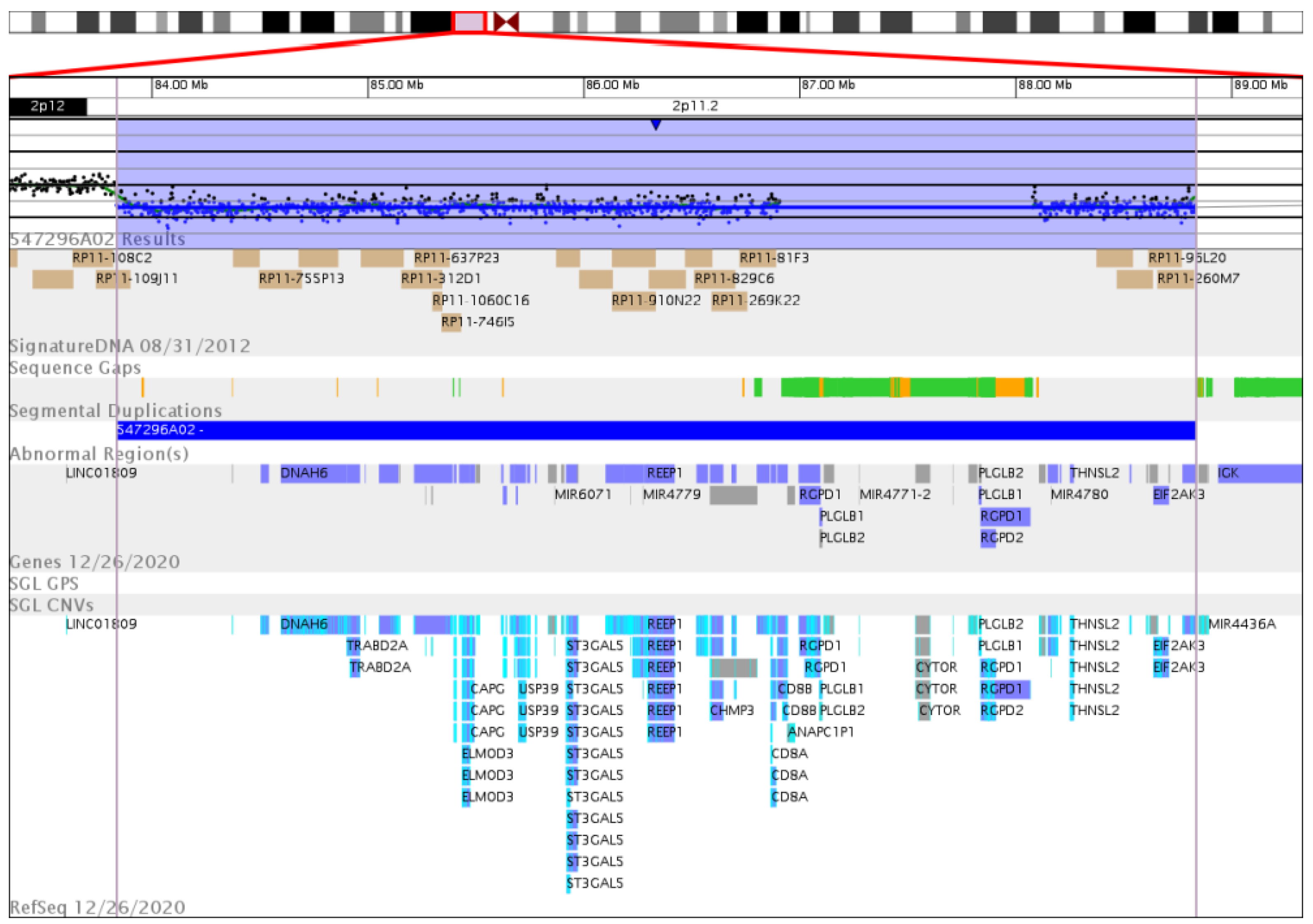
3.2. Genetic Analysis
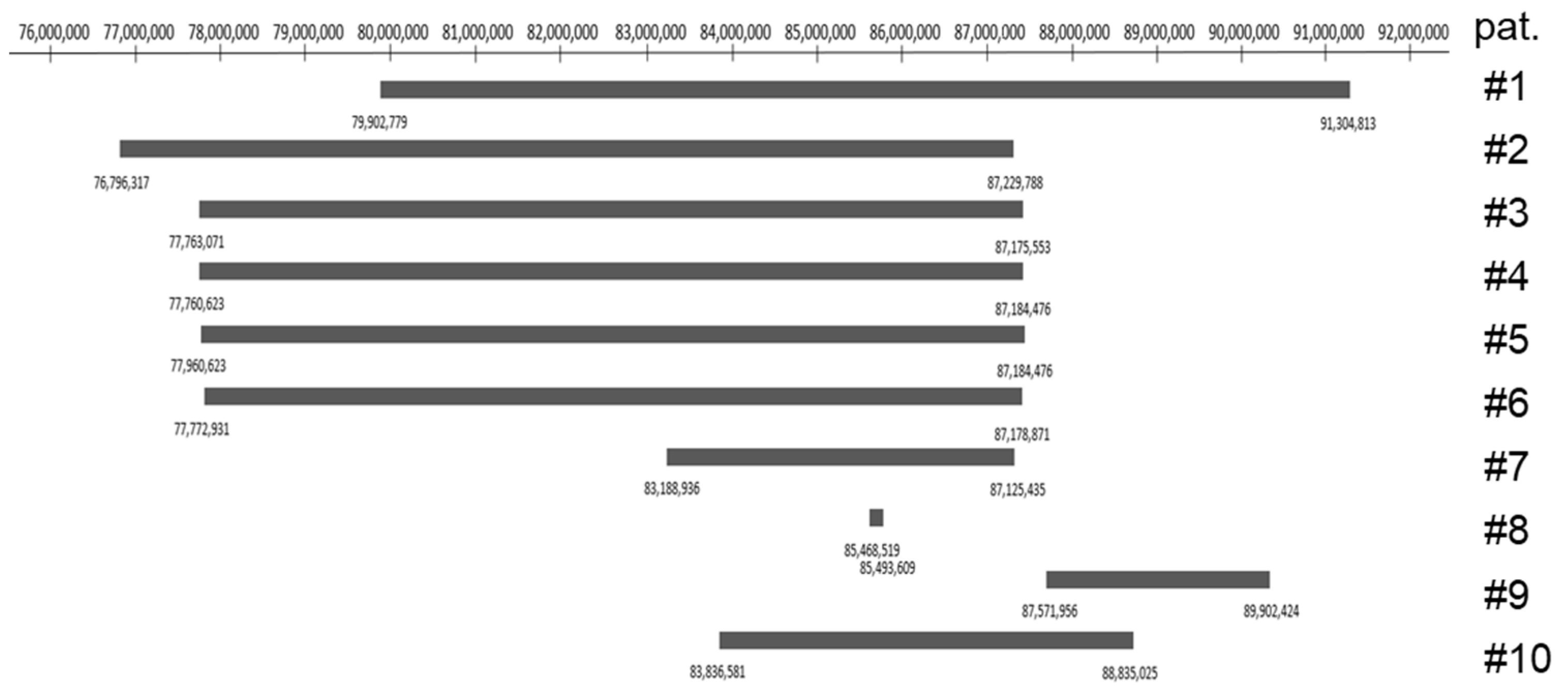
3.3. Literature Search
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McKusick, V.A. Mendelian Inheritance in Man and its online version, OMIM. Am. J. Hum. Genet. 2007, 80, 588–604. [Google Scholar] [CrossRef] [PubMed]
- Morley, T.J.; Han, L.; Castro, V.M.; Morra, J.; Perlis, R.H.; Cox, N.J.; Bastarache, L.; Ruderfer, D.M. Phenotypic signatures in clinical data enable systematic identification of patients for genetic testing. Nat. Med. 2021, 27, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Tzschach, A.; Graul-Neumann, L.M.; Konrat, K.; Richter, R.; Ebert, G.; Ullmann, R.; Neitzel, H. Interstitial deletion 2p11.2-p12: Report of a patient with mental retardation and review of the literature. Am. J. Med. Genet. Part A 2009, 149, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Writzl, K.; Lovrecić, L.; Peterlin, B. Interstitial deletion 2p11.2-p12: Further delineation. Am. J. Med. Genet. Part A 2009, 149, 2324–2326. [Google Scholar] [CrossRef] [PubMed]
- Rocca, M.S.; Fabretto, A.; Faletra, F.; Carlet, O.; Skabar, A.; Gasparini, P.; Pecile, V. Contribution of SNP arrays in diagnosis of deletion 2p11.2-p12. Gene 2012, 492, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Stevens, S.J.; Blom, E.W.; Siegelaer, I.T.; Smeets, E.E. A recurrent deletion syndrome at chromosome bands 2p11.2-2p12 flanked by segmental duplications at the breakpoints and including REEP1. Eur. J. Hum. Genet. 2015, 23, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Tassano, E.; Jagannathan, V.; Drögemüller, C.; Leoni, M.; Hytönen, M.K.; Severino, M.; Gimelli, S.; Cuoco, C.; Di Rocco, M.; Sanio, K.; et al. Congenital aural atresia associated with agenesis of internal carotid artery in a girl with a FOXI3 deletion. Am. J. Med. Genet. Part A 2015, 167, 537–544. [Google Scholar] [CrossRef]
- Silipigni, R.; Cattaneo, E.; Baccarin, M.; Fumagalli, M.; Bedeschi, M.F. Rare interstitial deletion of chromosome 2p11.2p12. Report of a new patient with developmental delay and unusual clinical features. Eur. J. Med. Genet. 2016, 59, 39–42. [Google Scholar] [CrossRef]
- Lahbib, S.; Leblond, C.S.; Hamza, M.; Regnault, B.; Lemée, L.; Mathieu, A.; Jaouadi, H.; Mkaouar, R.; Youssef-Turki, I.B.; Belhadj, A.; et al. Homozygous 2p11.2 deletion supports the implication of ELMOD3 in hearing loss and reveals the potential association of CAPG with ASD/ID etiology. J. Appl. Genet. 2019, 60, 49–56. [Google Scholar] [CrossRef]
- Baviera-Muñoz, R.; Martínez-Rubio, D.; Sastre-Bataller, I.; Campins-Romeu, M.; Losada-López, M.; Pérez-García, J.; Novella-Maestre, E.; Martínez-Torres, I.; Espinós, C. A 3.9-Mb Deletion on 2p11.2 Comprising the REEP1 Gene Causes Early-Onset Atypical Parkinsonism. Neurol. Genet. 2021, 7, e642. [Google Scholar] [CrossRef]
- Weaver, K.N.; Watt, K.E.; Hufnagel, R.B.; Navajas Acedo, J.; Linscott, L.L.; Sund, K.L.; Bender, P.L.; König, R.; Lourenco, C.M.; Hehr, U.; et al. Acrofacial Dysostosis, Cincinnati Type, a Mandibulofacial Dysostosis Syndrome with Limb Anomalies, Is Caused by POLR1A Dysfunction. Am. J. Hum. Genet. 2015, 96, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, K.; Watt, K.E.N.; Ide, S.; Baltrunaite, K.; Brunswick, C.; Inskeep, K.; Capannari, C.; Adam, M.P.; Begtrup, A.; Bertola, D.R.; et al. POLR1A variants underlie phenotypic heterogeneity in craniofacial, neural, and cardiac anomalies. Am. J. Hum. Genet. 2023, 110, 809–825. [Google Scholar] [CrossRef] [PubMed]
- Jaworek, T.J.; Richard, E.M.; Ivanova, A.A.; Giese, A.P.; Choo, D.I.; Khan, S.N.; Riazuddin, S.; Kahn, R.A.; Riazuddin, S. An alteration in ELMOD3, an Arl2 GTPase-activating protein, is associated with hearing impairment in humans. PLoS Genet. 2013, 9, e1003774. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sun, J.; Ling, J.; Li, J.; He, C.; Liu, Y.; Chen, H.; Men, M.; Niu, Z.; Deng, Y.; et al. ELMOD3, a novel causative gene, associated with human autosomal dominant nonsyndromic and progressive hearing loss. Hum. Genet. 2018, 137, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Feng, Y.; Chen, A.; Li, T.; Huang, S.; Liu, J.; Liu, X.; Liu, Y.; Gao, J.; Yan, D.; et al. Elmod3 knockout leads to progressive hearing loss and abnormalities in cochlear hair cell stereocilia. Hum. Mol. Genet. 2019, 28, 4103–4112. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wen, J.; Liu, X.; Chen, A.; Li, S.; Liu, J.; Sun, J.; Gong, W.; Kang, X.; Feng, Z.; et al. Gene regulation analysis of patient-derived iPSCs and its CRISPR-corrected control provides a new tool for studying perturbations of ELMOD3 c.512A>G mutation during the development of inherited hearing loss. PLoS ONE 2023, 18, e0288640. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Borel, C.; Ansar, M.; Jolly, A.; Makrythanasis, P.; Froehlich, C.; Iwaszkiewicz, J.; Wang, B.; Xu, X.; Li, Q.; et al. FOXI3 pathogenic variants cause one form of craniofacial microsomia. Nat. Commun. 2023, 14, 2026. [Google Scholar] [CrossRef]
- Quiat, D.; Timberlake, A.T.; Curran, J.J.; Cunningham, M.L.; McDonough, B.; Artunduaga, M.A.; DePalma, S.R.; Duenas-Roque, M.M.; Gorham, J.M.; Gustafson, J.A.; et al. Damaging variants in FOXI3 cause microtia and craniofacial microsomia. Genet. Med. Off. J. Am. Coll. Med. Genet. 2023, 25, 143–150. [Google Scholar] [CrossRef]
- Züchner, S.; Wang, G.; Tran-Viet, K.N.; Nance, M.A.; Gaskell, P.C.; Vance, J.M.; Ashley-Koch, A.E.; Pericak-Vance, M.A. Mutations in the novel mitochondrial protein REEP1 cause hereditary spastic paraplegia type 31. Am. J. Hum. Genet. 2006, 79, 365–369. [Google Scholar] [CrossRef]
- Beetz, C.; Pieber, T.R.; Hertel, N.; Schabhüttl, M.; Fischer, C.; Trajanoski, S.; Graf, E.; Keiner, S.; Kurth, I.; Wieland, T.; et al. Exome sequencing identifies a REEP1 mutation involved in distal hereditary motor neuropathy type V. Am. J. Hum. Genet. 2012, 91, 139–145. [Google Scholar] [CrossRef]
- Beetz, C.; Schüle, R.; Deconinck, T.; Tran-Viet, K.N.; Zhu, H.; Kremer, B.P.; Frints, S.G.; van Zelst-Stams, W.A.; Byrne, P.; Otto, S.; et al. REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain 2008, 131, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- de Bot, S.T.; van de Warrenburg, B.P.; Kremer, H.P.; Willemsen, M.A. Child neurology: Hereditary spastic paraplegia in children. Neurology 2010, 75, e75–e79. [Google Scholar] [CrossRef] [PubMed]
- Toft, A.; Birk, S.; Ballegaard, M.; Dunø, M.; Hjermind, L.E.; Nielsen, J.E.; Svenstrup, K. Peripheral neuropathy in hereditary spastic paraplegia caused by REEP1 variants. J. Neurol. 2019, 266, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Abreu, J.G.; Yokota, C.; MacDonald, B.T.; Singh, S.; Coburn, K.L.; Cheong, S.M.; Zhang, M.M.; Ye, Q.Z.; Hang, H.C.; et al. Tiki1 is required for head formation via Wnt cleavage-oxidation and inactivation. Cell 2012, 149, 1565–1577. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; An, Q.; Xi, H.; Yang, X.J.; Zhang, X.; Yuan, S.; Wang, J.; Hu, Y.; Liu, Q.; Fan, G. Single-Cell RNA Sequencing of hESC-Derived 3D Retinal Organoids Reveals Novel Genes Regulating RPC Commitment in Early Human Retinogenesis. Stem Cell Rep. 2019, 13, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Moise, A.R.; Lobo, G.P.; Erokwu, B.; Wilson, D.L.; Peck, D.; Alvarez, S.; Domínguez, M.; Alvarez, R.; Flask, C.A.; de Lera, A.R.; et al. Increased adiposity in the retinol saturase-knockout mouse. FASEB J. 2010, 24, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Chona, F.R.; Khan, A.N.; Chan, C.K.; Moore, A.N.; Dash, P.K.; Hernandez, M.R.; Lu, L.; Chesler, E.J.; Manly, K.F.; Williams, R.W.; et al. Genetic networks controlling retinal injury. Mol. Vis. 2005, 11, 958–970. [Google Scholar]
- Okubo, Y.; Masuyama, R.; Iwanaga, A.; Koike, Y.; Kuwatsuka, Y.; Ogi, T.; Yamamoto, Y.; Endo, Y.; Tamura, H.; Utani, A. Calcification in dermal fibroblasts from a patient with GGCX syndrome accompanied by upregulation of osteogenic molecules. PLoS ONE 2017, 12, e0177375. [Google Scholar] [CrossRef]
- Diñeiro, M.; Capín, R.; Cifuentes, G.; Fernández-Vega, B.; Villota, E.; Otero, A.; Santiago, A.; Pruneda, P.C.; Castillo, D.; Viejo-Díaz, M.; et al. Comprehensive genomic diagnosis of inherited retinal and optical nerve disorders reveals hidden syndromes and personalized therapeutic options. Acta Ophthalmol. 2020, 98, e1034–e1048. [Google Scholar] [CrossRef]
- Borna, N.N.; Kishita, Y.; Kohda, M.; Lim, S.C.; Shimura, M.; Wu, Y.; Mogushi, K.; Yatsuka, Y.; Harashima, H.; Hisatomi, Y.; et al. Mitochondrial ribosomal protein PTCD3 mutations cause oxidative phosphorylation defects with Leigh syndrome. Neurogenetics 2019, 20, 9–25. [Google Scholar] [CrossRef]
- Huck, J.H.; Verhoeven, N.M.; Struys, E.A.; Salomons, G.S.; Jakobs, C.; van der Knaap, M.S. Ribose-5-phosphate isomerase deficiency: New inborn error in the pentose phosphate pathway associated with a slowly progressive leukoencephalopathy. Am. J. Hum. Genet. 2004, 74, 745–751. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age (years) | 4 | 6 | 8 | 12 |
|---|---|---|---|---|
| Distance visual acuity | OU: 0.63 | OD: 0.16 OS: 0.2 | OD: 0.5 OS: 0.63 | OU: 1.0 |
| Near visual acuity | OU: 0.5 | OD: 0.16 | OD: 0.63 OS: 0.25 | OD: 0.4 OS: 0.5 |
| Gene | Gene Product | Inheritance | Disease | OMIM |
|---|---|---|---|---|
| FUNDC2P2 | FUN14 domain containing 2 pseudogene 2 | |||
| SUCLG1 | Succinate-CoA Ligase, GDP/ADP-forming, α subunit | AR | Mitochondrial DNA depletion syndrome 9 | 245400 |
| DNAH6 | Dynein, axonemal, heavy chain 6 | |||
| TRABD2A | TRAB domain-containing protein 2a | |||
| TMSB10 | Thymosin, β-10 | |||
| KCMF1 | Potassium channel modulatory factor 1 | |||
| LINC01964 | Long intergenic non-protein coding RNA 1964 | |||
| TCF7L1-IT1 | TCF7L1 intronic transcript 1 | |||
| TCF7L1 | Transcription factor 7-like 1 | |||
| LOC102724579 | ||||
| TGOLN2 | Trans-Golgi network protein 2 | |||
| RETSAT | Retinol saturase | |||
| ELMOD3 | ELMO/CED12 domain-containing protein 3 | AD AR | Deafness 81 Deafness 88 | 619500 615429 |
| CAPG | Capping protein, gelsolin-like | |||
| SH2D6 | SH2 domain containing 6 | |||
| PARTICL | Promoter of MAT2 AS radiation-induced circulating lncRNA | |||
| MAT2A | Methionine adenosyltransferase 2A | |||
| GGCX | γ-glutamyl carboxylase | AR | Combined deficiency of Vitamin K-dependent clotting factors Pseudoxanthoma elasticum-like disorder withmultiple coagulation factor deficiency | 277450 610842 |
| VAMP8 | Vesicle-associated membrane protein 8 | |||
| VAMP5 | Vesicle-associated membrane protein 5 | |||
| RNF181 | Ring finger protein 181 | |||
| TMEM150A | Transmembrane protein 150a | |||
| USP39 | Ubiquitin-specific protease 39 | |||
| C2ORF68 | Chromosome 2 open reading frame 68 | |||
| SFTPB | Surfactant protein B | AR | Pulmonary surfactant metabolism dysfunction 1 | 265120 |
| GNLY | Granulysin | |||
| ATOH8 | Atonal-homolog 8 | |||
| MIR6071 | MicroRNA 6071 | |||
| LOC284950 | ||||
| ST3GAL5 | ST3 β-galactoside α-2,3-sialyltransferase 5 | AR | Salt and pepper developmental regression syndrome | 609056 |
| ST3GAL5-AS1 | ST3GAL5 antisense RNA 1 | |||
| POLR1A | RNA polymerase I subunit A | AD | Acrofacial dysostosis, Cincinnati type | 616462 |
| PTCD3 | Pentatricopeptide repeat domain 3 | AR | Combined oxidative phosphorylation deficiency 51 | 619057 |
| SNORD94 | Small nucleolar RNA, C/D box 94 | |||
| IMMT | Inner membrane mitochondrial protein | |||
| MIR4779 | MicroRNA 4779 | |||
| MRPL35 | Mitochondrial ribosomal protein L35 | |||
| REEP1 | Receptor expression-enhancing protein 1 | AD AR | Spastic paraplegia 31 Distal hereditary motor neuronopathy 12 Distal hereditary motor neuronopathy 6 | 610250 614751 620011 |
| KDM3A | Lysine-specific demethylase 3a | |||
| CHMP3 | Charged multivesicular body protein 3 | |||
| RNF103 | Ring finger protein 103 | |||
| RMND5A | Required for meiotic nuclear division 5 homolog a | |||
| CD8A | CD8 antigen, α polypeptide | AR | Familial CD8 deficiency | 608957 |
| CD8B | CD8 antigen, β polypeptide | |||
| ANAPC1P1 | ANAPC1 pseudogene 1 | |||
| RGPD1 | RANBP2 like and GRIP domain containing 1 | |||
| RGPD2 | RANBP2 like and GRIP domain containing 2 | |||
| PLGLB2 | Plasminogen like B2 | |||
| PLGLB1 | Plasminogen like B1 | |||
| LOC285074 | ||||
| MIR4771-2 | MicroRNA 4771-2 | |||
| MIR4772-1 | MicroRNA 4771-1 | |||
| CYTOR | Cytoskeleton regulator RNA | |||
| MIR4435-1 | MicroRNA 4435-1 | |||
| MIR4435-2 | MicroRNA 4435-2 | |||
| KRCC1 | Lysine rich coiled-coil 1 | |||
| FABP1 | Fatty acid binding protein 1 | |||
| SMYD1 | SET and MYND domain-containing protein 1 | |||
| MIR4780 | MicroRNA 4780 | |||
| THNSL2 | Threonine synthase like 2 | |||
| FOXI3 | Forkhead box I3 | AD/AR | Craniofacial microsomia 2 | 620444 |
| TEX37 | Sperm microtubule inner protein 9 | |||
| LOC101928371 | ||||
| EIF2AK3 | Eukaryotic translation initiation factor 2 α kinase 3 | AR | Wolcott–Rallison syndrome | 226980 |
| LOC101928403 | ||||
| RPIA | Ribose 5-phosphate isomerase A | AR | Ribose 5-phosphate isomerase deficiency | 608611 |
| ANKRD36BP2 | Ankyrin repeat domain 36B pseudogene 2 | |||
| MIR4436A | MicroRNA 4436a |
| Patient No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Tzschach et al., 2009 [3] | Writzl et al., 2009 [4] | Rocca et al., 2012 [5] | Stevens et al., 2015 [6] | Stevens et al., 2015 [6] | Silipigni et al., 2015 [8] | Baviera-Munoz et al., 2021 [10] | Lahbib et al., 2019 [9] | Tassano et al., 2015 [7] | Present Case |
| Position in 2p | p11.2p12 | p11.2p12 | p11.2p12 | p11.2p12 | p11.2p12 | p11.2p12 | p11.2 | p11.2 | p11.2 | p11.2 |
| Deletion size | 11.4 Mb | 10.4 Mb | 9.2 Mb | 9.4 Mb | 9.4 Mb | 9.4 Mb | 3.9 Mb | 25.09 kb | 2.4 Mb | 5.0 Mb |
| Genomic Localization | 79,902,77–91,304,813 | 76,796,317–87,229,788 | 77,763,071–87,175,553 | 77,760,623–87,184,476 | 77,760,623–87,184,476 | 77,772,931–87,178,871 | 83,188,936–87,125,435 | 85,468,519–85,493,609 | 87,571,956–89,902,424 | 83,836,581–88,835,025 |
| Sex | F | M | F | M | M | F | F | M | F | M |
| Inheritance | de novo | de novo | de novo | de novo | de novo | de novo | de novo | parental, homozygous | paternal | de novo |
| Age evaluation | 5 y | 5 y 9 m | 9 y | 15 y 8 m | 5 y 4 m | 12 m | 35 y | 7 y | 4 m | 11 y 9 m |
| Microcephaly | + | - | - | - | - | - | - | - | - | - |
| High forehead | + | + | + | + | + | + | + | - | - | - |
| Broad high nasal bridge | + | + | + | + | + | + | (+) | - | - | + |
| Ear anomalies | + | + | + | + | + | + | + | + | + | + |
| Feet anomalies | + | + | - | + | - | - | - | - | - | + |
| Digital anomalies | + | + | - | - | - | - | - | - | - | - |
| Growth retardation | + | + | + | - | - | - | + | - | - | - |
| Speech delay | + | + | + | + | + | n.a. | - | + | n.a. | - |
| Delayed motor development | + | + | + | + | + | + | + | + | - | + |
| Hypertonia | - | + | - | hypotonia | - | - | - | hypotonia | - | hypotonia |
| Ataxia | - | + | + | - | - | - | - | - | - | - |
| Intellectual disability | + | mild | mild | moderate | borderline | mild | - | + | ? | + |
| Happy disposition | + | + | - | + | + | + | ? | - | ? | + |
| Other symptoms | single umbilical artery | vesico-ureteral reflux | incomplete myelination white matter, hearing impairment | hyperreflexia of lower limbs, clumsy gait | hypermobile hands | bilateral choanal atresia, atrial septal defect | atypical early-onset parkinsonsim with dystonia and lower limb spasticity, scoliosis, strabismus, amblyopia | autism, hearing impairment, primary enuresis | congenital aural atresia, agenesis of internal carotid artery, velopharyngeal insufficiency, aberrant course of left facial nerve, microtia, hearing impairment | pendular horizontal nystagmus, visual acuity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrario, A.; Aliu, N.; Rieubland, C.; Vuilleumier, S.; Grabe, H.M.; Escher, P. Expanding Genotype/Phenotype Correlation in 2p11.2-p12 Microdeletion Syndrome. Genes 2023, 14, 2222. https://doi.org/10.3390/genes14122222
Ferrario A, Aliu N, Rieubland C, Vuilleumier S, Grabe HM, Escher P. Expanding Genotype/Phenotype Correlation in 2p11.2-p12 Microdeletion Syndrome. Genes. 2023; 14(12):2222. https://doi.org/10.3390/genes14122222
Chicago/Turabian StyleFerrario, Alessandra, Nijas Aliu, Claudine Rieubland, Sébastian Vuilleumier, Hilary M. Grabe, and Pascal Escher. 2023. "Expanding Genotype/Phenotype Correlation in 2p11.2-p12 Microdeletion Syndrome" Genes 14, no. 12: 2222. https://doi.org/10.3390/genes14122222
APA StyleFerrario, A., Aliu, N., Rieubland, C., Vuilleumier, S., Grabe, H. M., & Escher, P. (2023). Expanding Genotype/Phenotype Correlation in 2p11.2-p12 Microdeletion Syndrome. Genes, 14(12), 2222. https://doi.org/10.3390/genes14122222