1. Introduction
Normal coronary artery anatomy consists of the coronary arteries arising from the aortic sinuses, which follow a converging path toward the heart’s anatomical apex. The three main coronary arteries (the right coronary artery (RCA), left anterior descending (LAD), and left circumflex (LCX)) each have common placements and travel along the epicardium of the heart in patients who underwent typical coronary development [
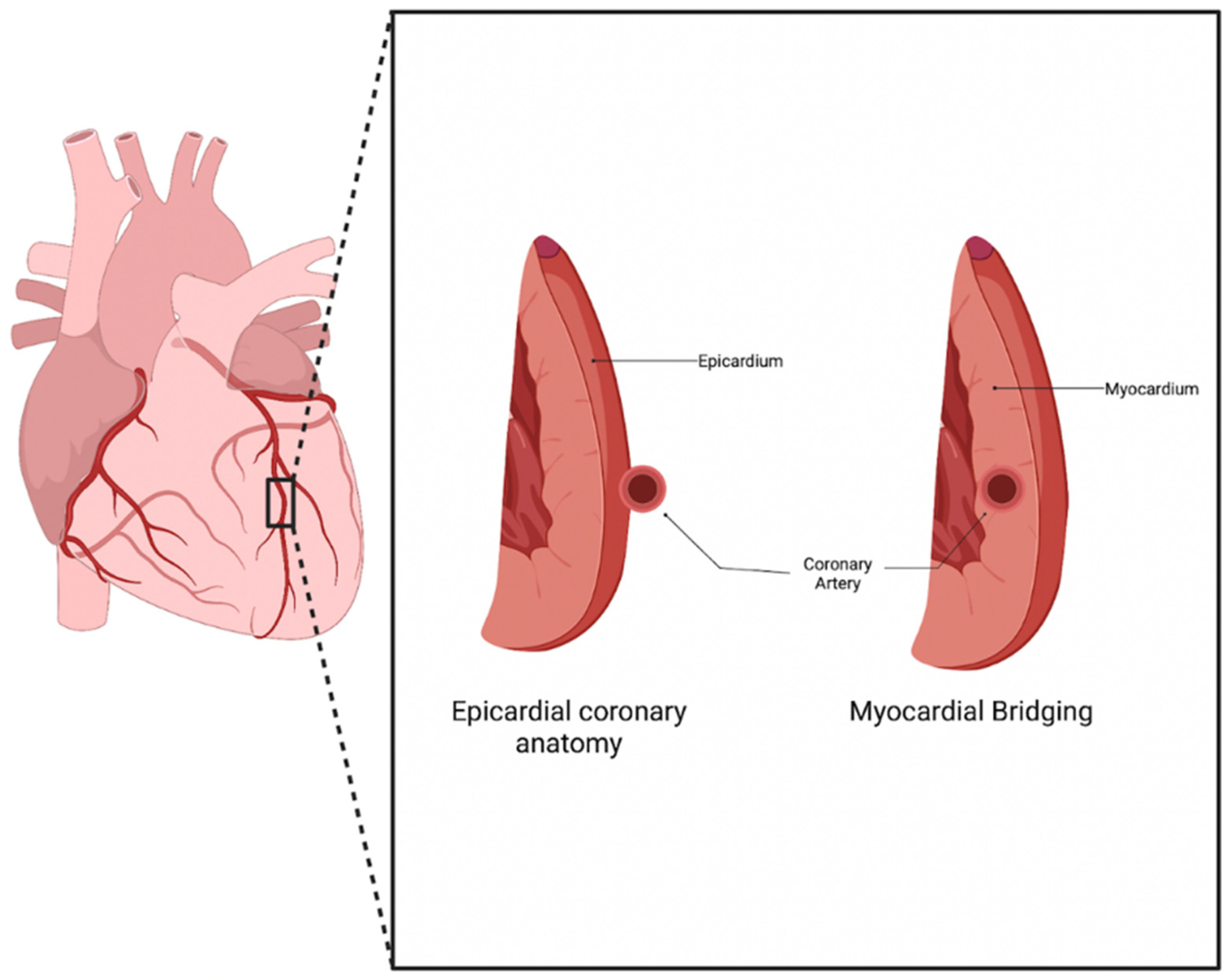
1]. Myocardial bridging (MB) is a congenital anomaly that consists of a portion of an epicardial coronary artery diving into the myocardium layer for a section of its journey (
Figure 1). The muscle layer that lies above the artery is recognized as a myocardial bridge, while the portion of the artery is labeled as a tunneled artery. This anomaly is typically seen in the LAD, but may also be present in any of the coronary arteries [
2]. Our clinical experience has verified this phenotypic presentation.
The genetic mechanisms driving the development of congenital coronary vascular anomalies (CCVAs), including those of myocardial bridging, lack sufficient research [
4]. This finding is surprising as these anomalies are present and well characterized in patients throughout the world. The rates of myocardial bridging vary between the methods of evaluation, with autopsy and angiography being the most common techniques used to assess for the presence of bridging. Invasive angiography generally underreports the prevalence of myocardial bridging, as it typically detects the anomaly in 0.15–25% of patients [
1]. This low prevalence is countered by autopsy reports, which illustrate the prevalence of MBs in 5–86% of patients [
5]. Recent computed tomography (CT) studies have also shown bridging in up to 25% of patients, further showing the underestimation of its prevalence within angiography studies [
1]. In our own CCTA cohort from 2014–2023, the prevalence is 7% (70/1000) [
6]. The discrepancy in these methods demonstrates the unreliability of the current diagnostic methods and suggests a need for improved techniques.
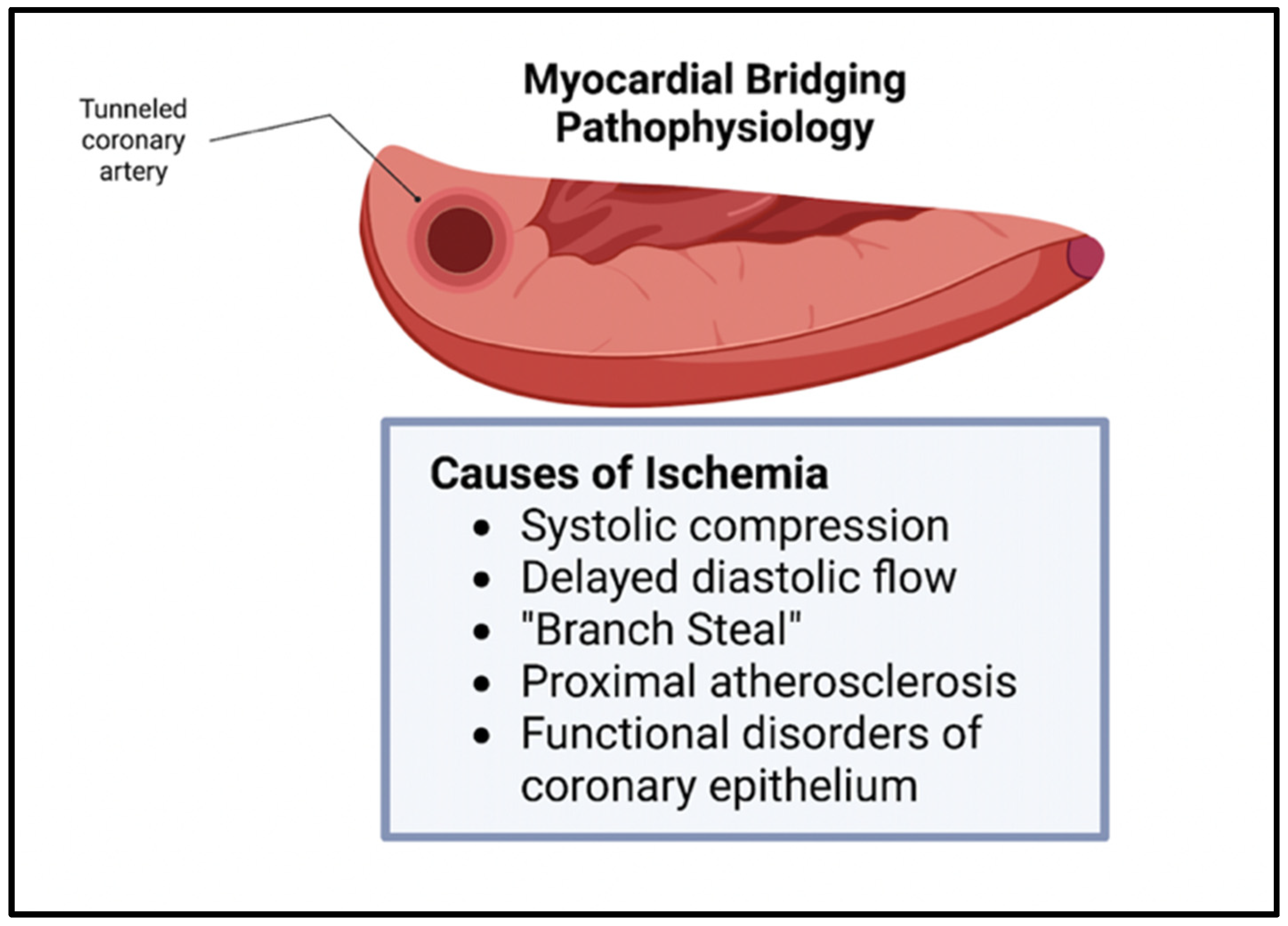
Though bridging was previously viewed as benign, this belief has been challenged in the recent literature. The vessel compression of the intramural artery has been noted during the contraction of the myocardium, often leading to chest pain and, sometimes, to acute coronary syndrome [
7]. The overarching pathophysiology behind these symptoms stems from the myocardial ischemia secondary to the tunneling and compression of the artery, with the ischemia being exacerbated by the increased sympathetic tone [
2]. Since the systolic phase of the myocardial cycle is only mildly involved in the perfusion of the myocardial tissue, other mechanisms contribute to the ischemia seen in bridging. These mechanisms include delayed early diastolic artery relaxation, atherosclerotic stenosis development proximal to the tunneled artery, functional disorders of coronary circulation including impaired endothelium-dependent vasodilatation and microvascular dysfunction, and the “branch steal” phenomenon (
Figure 2). The branch steal effect describes the crossing of blood through the constricted segment during the end of systole and early diastole, leading to an increase in diastolic flow velocity in the artery. The ischemia due to these various mechanisms are important causes of the anginal symptoms that bridging patients experience [
8]. Other cardiovascular events linked to myocardial bridging include cardiac arrhythmias and sudden cardiac death [
9].
Objectives
Myocardial bridging is a symptomatic cardiac anomaly that is underdiagnosed in many individuals and has historically been under-researched. A few systematic reviews have been published detailing its clinical features and hypothesizing mechanisms regarding myocardial bridge development throughout the last decade, however, research into the genomic association behind bridge development remains sparse. The objective of this manuscript is to review and discuss prior discoveries related to genomics and bridge development, serving as an up-to-date reference to prompt further hypothesis-driven investigations.
2. Methods
2.1. Overview
This study used a structured and comprehensive approach that included a methodological approach for capturing articles containing research on myocardial bridging and genomic variance. Various popular scientific databases were used to gather relevant articles, and strict inclusion and exclusion guidelines were followed by each author.
2.2. Published Literature Search
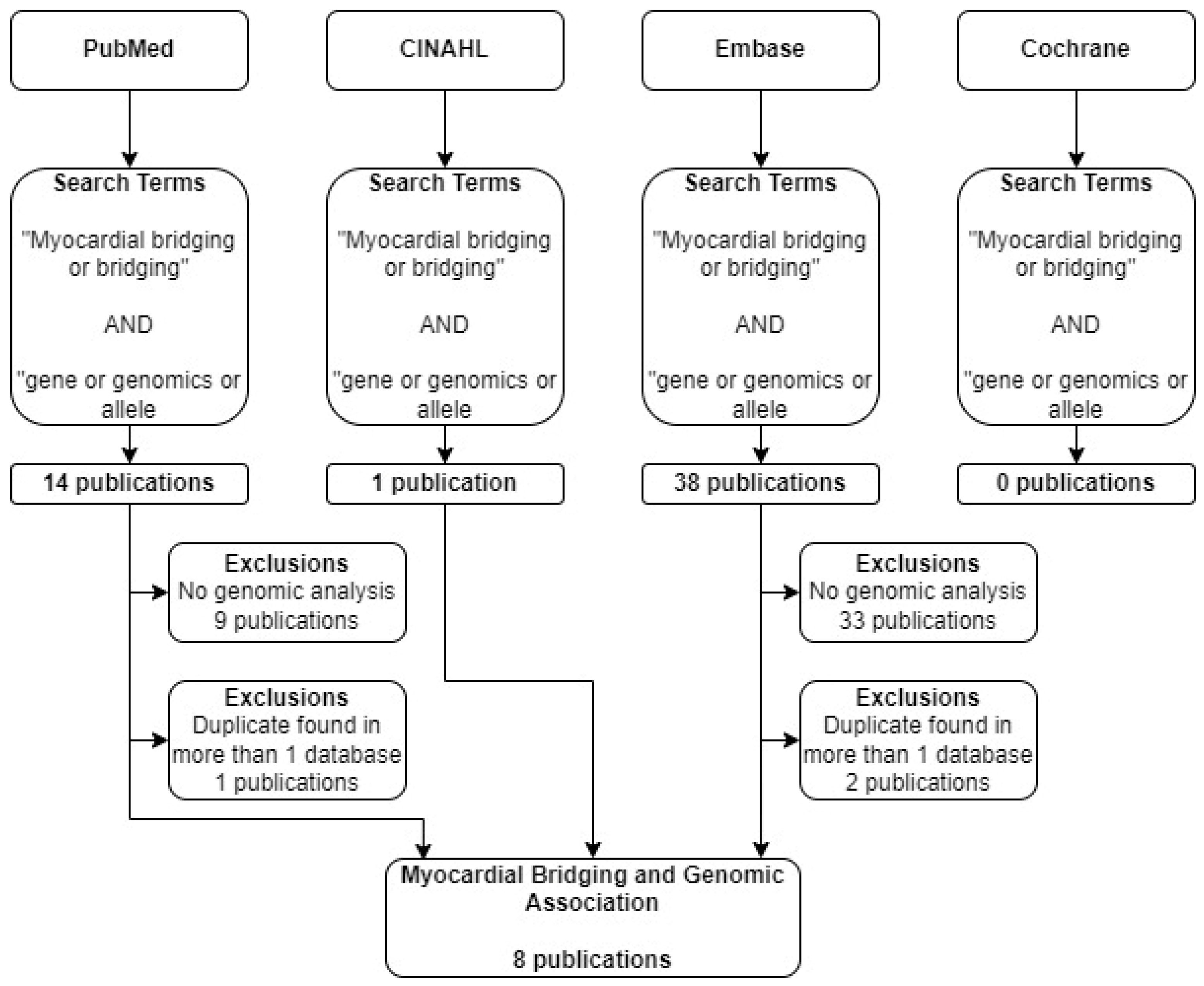
A literature search was conducted using Pubmed, CINAHL, EMBASE, and Cochrane (
Table 1). The search terms used in each of these databases are summarized in
Figure 1. The terms “myocardial bridging” and “genomics” were searched in each of the listed databases. Only original, peer-reviewed, clinical research studies published in English within the last 10 years were considered. A search of the Cochrane database yielded no relevant results. Out of the articles included, several were found in more than one of the databases searched.
For inclusion in our study, the following inclusion criteria had to be met:
Published on or after 1 January 2013.
Study published in English.
Evidence of discussion on myocardial bridging and its association with a gene, allele variant, or genomic association.
Studies were excluded from further consideration if the exclusion criteria were met including:
Study published prior to 1 January 2013.
Study not published in English.
No mention of an association between myocardial bridging and genomics or genetic variance.
2.3. Data Extraction and Analysis
Each author conducted a search in Pubmed, CINAHL, EMBASE, and Cochrane. All authors yielded the same number of results. Out of the resultant papers, each author identified which papers to include. Each author yielded an identical list, representing congruence amongst the authors. Once the resultant papers were reviewed by each author and found to have met the inclusion criteria, a final list of resultant articles was synthesized.
From the 53 papers found across our four databases, we extracted relevant study characteristic, genomic, and biologic data from our resultant eight papers. These data included the cause of death, mutation, and subject information. These clinical and genomic variables are detailed below (
Table 2).
Study Characteristics
A total of 53 eligible articles were found using our search criteria. Out of these, eight met our inclusion criteria and involved the discussion of myocardial bridging and genomic analysis (
Figure 3). Out of these, 6 were case reports (
Table 2). Each of these articles and case reports were read by each author and discussed in detail.
2.4. Case Reports
Out of the eight resultant papers that met our inclusion criteria, six were case reports. The mean subject age was 18 and the median age 15.5. Four out of six (4/6, 66%) of the subjects were female. Two of the six (2/6, 33%) papers discussed an MYH7 mutation. The remaining four papers reviewed other genes. Two of the six (2/6, 33%) subjects died of sudden cardiac death.
2.5. Research Articles
Out of the eight resultant papers that met our inclusion criteria, two were research articles. The first discussed the microRNA expression profile seen in myocardial bridge patients. The second paper discussed the cardiac manifestations of the PRKAG2 mutation within the context of a case presentation.
3. Genetic Mutations Associated Myocardial Bridges: Potential Candidate Genes?
3.1. MYH7
ß-myosin heavy chain (
MYH7) gene mutations have been associated with hypertrophic cardiomyopathy (HCM) and restrictive cardiomyopathy (RCM). The
MYH7 mutations found in a 9-month-old female with RCM and associated myocardial hypertrophy and a 7-year-old female who died of sudden cardiac death (SCD) were classified as variants of unknown significance (VUS). Of note, both patients were found to have myocardial bridging of the left anterior descending (LAD) artery on autopsy [
10,
11]. Genetic testing of one of the patients revealed an A1157G mutation in exon 13 of the
MYH7 gene. This mutation resulted in a nonsynonymous amino acid change at position 386 from tyrosine to cysteine and was classified as being likely pathogenic [
10,
11].
The
MYH7 gene encodes for ß-myosin heavy chain, an essential component of the cardiac sarcomere. Though the authors do not hypothesize a mechanism regarding bridge formation, there are other examples of associations between
MYH7 mutations and bridge formation including the case of a 15-year-old with exertional dyspnea and chest pain [
12]. We view this as a potential candidate gene of interest for further studies, as mutations in the gene may have significant clinical manifestations outside of its already known associations with HCM and RCM.
3.2. DPP6
The
DPP6 gene encodes a membrane protein that is a member of the peptidase S9B family of serine proteases and is known to bind specific voltage-gated potassium channels [
13]. Mutations in the
DPP6 gene have been previously associated with the development of idiopathic ventricular fibrillation (IVF), which is known to cause SCD [
14]. Mutations in the
DPP6 (Ala714Thr) gene were discovered in the father and sister of a child who died from SCD and was found to have a 1.1 × 0.5 cm myocardial bridge on autopsy. Further analysis of
DPP6 and the other mutated genes in the family of the proband found that they were variants of unknown significance (VUS). Importantly, the
DPP6 mutation was not the only mutation in the child. The VUS in
MYH7, SCN2B, and
NOTCH1 were also found. The authors hypothesize that the SCD seen in the child was likely secondary to the HCM and its subsequent role in arrhythmic death, however, they are unsure of the consequences secondary to the isolated genetic variants [
11]. We suspect that the
DPP6 mutation may have contributed to arrhythmias seen in the patient, however, we have low suspicion that the mutation played a role in bridge formation.
3.3. SCN2B
The
SCN2B gene encodes voltage-gated sodium channel ß2-subunits, and is involved in cell–cell adhesion and cell migration [
15]. Mutations in the
SCN2B gene have been associated with cardiac arrhythmias in humans including atrial fibrillation and Brugada syndrome [
16]. Mutations in the
SCN2B gene (Glu31Asp) were discovered in a child that died from sudden cardiac death who was found to have a myocardial bridge on autopsy [
11]. No mechanism regarding the gene mutation and the development of myocardial bridging was discussed in the report. Despite this, we hypothesize that the disruption in
SCN2B’s known role in cell migration via genetic mutation may play a role in the abnormal migration of the coronary arteries during embryogenesis, which could subsequently result in the formation of myocardial bridging. Due to its known role in this vital process, further investigation into the potential relationship between mutations in
SCN2B and bridge development may be promising.
3.4. NOTCH1
NOTCH1 encodes a cell-surface receptor that plays a role in the development of various cell and tissue types [
17]. The importance of the NOTCH pathway during the development of the cardiovascular system has been extensively discussed. Notably, NOTCH has several roles during ventricular development including that of trabecular development via differentiation and the proliferation of cardiomyocytes [
18]. A mutation in
NOTCH1 (Arg2313Gln) was discovered in a young female that died from SCD who was found to have a myocardial bridge on autopsy [
11]. Though the authors made no direct hypothesis regarding a link between
NOTCH1 and bridge development, it is plausible that mutations in a member of the NOTCH family of genes, known players in cardiovascular development, may play a role in the development of a coronary artery anomaly such as myocardial bridging.
Studies have demonstrated that
NOTCH1 plays a pivotal role in cardiomyocyte differentiation and proliferation. The gene has been implicated in various congenital heart defects including hypoplastic left heart syndrome [
19]. The core of
NOTCH1’s role in angiogenesis is its impact on endothelial cells. The gene influences the delicate balance between tip cells and stalk cells during sprouting angiogenesis. Tip cells, located at the leading edge of growing vessels, steer vessel extension, while stalk cells support vessel elongation.
NOTCH1 activity in tip cells curtails excessive sprouting by inducing the expression of Dll4, a NOTCH ligand. This creates a feedback loop where Dll4 interacts with the neighboring endothelial cells’ NOTCH receptors, repressing their tip cell potential and promoting stalk cell characteristics, thereby maintaining vessel integrity [
20]. A disruption in this balance may result in a disruption of angiogenesis.
NOTCH1 is activated in proliferating embryonic and immature cardiomyocytes and is downregulated in the myocardium during postnatal development. NOTCH signaling in adults is activated transiently in response to myocardial injury, further suggesting that the gene is contributory to cardiac repair via angiogenesis [
21,
22]. Due to the many roles that the
NOTCH1 gene plays in the angiogenesis and development of the cardiovascular system, further investigation assessing its potential association in bridge development is warranted as we view
NOTCH1 to be a candidate gene of interest.
3.5. SLMAP
A variant of unknown significance in the
SLMAP gene (c.599c>T) was found in a 20-year-old male that passed away from sudden cardiac death. Further autopsy revealed a 2-cm myocardial bridge that began in the left anterior descending artery. The missense mutation was found to have a very low frequency in the general population, and the mutation was found to be likely pathogenic using three in silico predictors [
23].
SLMAP is a known cardiac membrane protein that plays a role in excitation–contraction coupling in cardiac myocytes.
SLMAP mutations have been previously linked to cardiac conditions including the development of heart failure [
24].
SLMAP has also been associated with the development of Brugada syndrome, a cardiac channelopathy, through the silencing of
SLMAP by small-interfering RNA (siRNA) [
25]. No relationship between
SLMAP and angiogenesis, cell migration, or coronary artery embryogenesis were discussed, limiting our suspicion for its involvement in myocardial bridge development. Based on
SLMAP’s role in cardiac ion channel physiology rather than processes vital to coronary artery development and maturation, we do not view this as a potential candidate gene for further studies.
3.6. COMMD10
Deletions in
COMMD10 were identified in a 19-year-old athlete who was experiencing syncopal episodes. The coronary CTA of the patient discovered a 20 mm bridge in the LAD. An in-depth genetic analysis was performed using array-comparative genomic hybridization (array-CGH) and whole exome sequencing. The array-CGH demonstrated a copy number variation (CNV) alteration affecting intron 5 of the
COMMD10 gene, which is responsible for modulating the activity of the cullin-RING E3 ubiquitin ligase complexes (CRL) and reducing NFK-ß activation.
COMMD10 has also been found to be expressed in the endothelial cells and smooth muscle cells of various tissues, which demonstrates a potential involvement in angiogenesis during stages of embryogenesis. The gene has also been found to be expressed in cardiac tissue, which suggests it may have a role in cardiac development. The authors of the study hypothesize that
COMMD10 plays an important role during cardiac development, which may include the development of congenital heart disorders and anomalies such as myocardial bridging [
26]. Due to these findings, we agree that
COMMD10 is a candidate gene of interest and believe that further research regarding the association of the gene with myocardial bridge development is warranted.
3.7. MACROD2
MACROD2 is a deacetylase involved in removing ADP-ribose from mono-ADP-ribosylated proteins and is frequently involved in patients with complex syndromes [
27]. A macrodeletion of
MACROD2 was discovered in an athlete suffering from syncope who was found to have a 20 mm myocardial bridge. An array-CGH demonstrated CNVs, which may be associated with the presence of congenital heart disorders (CHD). A recent genome-wide association study that included over 4000 patients affected by CHD and 8000 controls revealed a statistically significant association between MACROD2 polymorphisms and the development of the transposition of the great vessels. Further data has shown the expression of
MACROD2 is present in human embryonic cardiac cells, further strengthening the possibility of the gene’s involvement as a transcriptional regulator in cardiomyocytes [
26]. Due to its presence in human embryonic cardiac cells and its potential role as a transcriptional regulator in cardiac myocytes, the authors hypothesized its contributing role in the development of CHDs. With these findings, we agree that this may be a candidate gene of interest and support further investigation into this genotype–phenotype relationship.
3.8. FBN1
A heterozygous mutation in the
FBN1 gene was found on whole exome sequencing in a 12-year-old girl that was experiencing recurrent syncopal episodes after exercise. The identified change was a missense mutation of c.3535A>G resulting in an amino acid change of p.I1175M in exon 29. Further bioinformatic analysis suggested that this variant may affect the structure and function of the protein product [
28]. The
FBN1 gene encodes the fibrillin-1 protein, an extracellular matrix component that regulates growth factor signaling pathways. Pathologic mutations in
FBN1 are well-established causes of Marfan syndrome and other congenital heart and vascular defects [
29]. There are no established associations between
FBN1 and coronary artery development, however, with the gene’s extensive role in other cardiovascular pathologies, genes in this cascade (FBN1 and FBN2) should be further characterized in myocardial bridge development as candidate genes of interest.
3.9. DES
A heterozygous missense variant (c.1300G>A; p.E434K) of the
DES gene was identified using whole-exome sequencing in a Chinese family with cardiomyopathy and sudden cardiac death. A myocardial bridge was discovered in the proband; however, none was present in the other four patients. Further genetic analysis predicted the mutation to be pathogenic, which was strengthened with its absence in 200 controls. The
DES gene encodes for desmin, an intermediate filament protein that stabilizes sarcomeres and cell contacts in the cardiac intercalated disc [
30]. We have little suspicion to believe that the isolated
DES mutation was the causative agent for bridge development. This belief is secondary to the lack of evidence of bridging found in the proband’s siblings or parents, or in other reports.
3.10. PRKAG2
The PRKAG2 gene is best known for its role in Wolff-Parkinson-White (WPW) and PRKAG2 syndrome. PRKAG2 syndrome is glycogen accumulation within cardiac tissue, which clinically presents as HCM. A mutation in the PRKAG2 gene was found in a 23-year-old female with a known history of WPW and HCM, who presented after a non-ST elevation myocardial infarction. The patient underwent genetic testing which revealed a heterozygous missense Arg302Gln mutation in the PRKAG2 gene. The angiography revealed severe bridging of the LAD in the patient [
31]. Though this mutation may have contributed to myocardial hypertrophy secondary to the glycogen accumulation, we do not view this as a likely candidate gene for bridging development. We believe that the associated myocardial bridge in this patient is likely secondary to the underlying PRKAG2 syndrome, and not due to a primary mutation causing abnormal coronary artery development or migration.
3.11. Summary of Genes
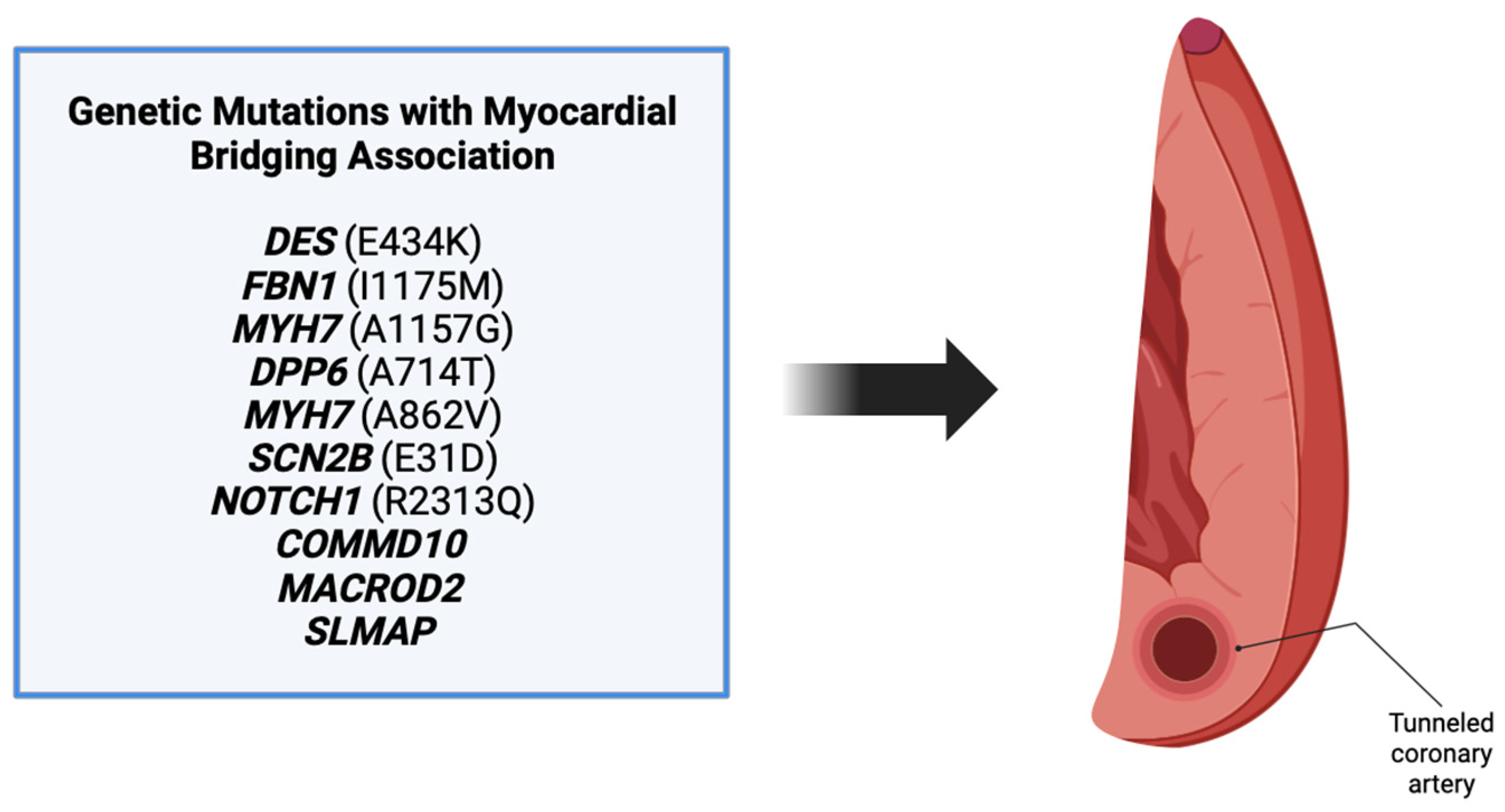
Our review revealed a total of 10 genes in patients that were also found to have a myocardial bridging phenotype (
Figure 4). Each of these genes have different roles in various processes including angiogenesis, embryogenesis, and cell differentiation, as discussed above. Patients in the cases varied in their presentation, with many being symptomatic and others having incidental findings of myocardial bridging on imaging or autopsy. Regardless of the presentation, the background of each gene was reviewed to determine its potential role in myocardial bridge development.
Of the mutations discovered in the review, we only view six to be candidate genes for further research:
MYH7,
SCN2B,
NOTCH1,
COMMD10,
MACROD2, and
FBN1/FBN2 (
Table 3). These genes have been found to be key in various processes that are vital for coronary artery development and/or migration, and it is plausible that disruptions in these genes may serve as a genesis for the development of myocardial bridging. These findings warrant further investigation into the potential mechanistic basis behind mutations in the candidate genes and the development of myocardial bridging.
4. Myocardial Bridging and MicroRNA Expression
MicroRNAs (miRNAs) are short, non-coding RNAs that act as post-transcriptional regulators. Prior studies have identified miRNAs as having tissue or cell-specificity, making them potential diagnostic tools. Of note, five circulating miRNAs have been identified as possible biomarkers for myocardial bridge detection. These miRNAs, miR-29b, miR-151-3p, miR-126, miR-503-3p, and miR-645, were all capable of distinguishing patients with known myocardial bridges from control patients [
32]. This is an important finding that needs further exploration through a larger cohort of genomic study in CT angiographically confirmed myocardial bridge patients. If positive findings are reported, these miRNAs may serve as novel genetic biomarkers for more cost-effective myocardial bridge screening. Furthermore, use of these biomarkers may improve diagnostic accuracy in patients with myocardial bridging, as current imaging resources vary in reliability. Due to this, further investigation into the relationship between various miRNAs and myocardial bridging presence is warranted.
5. Conclusions
Myocardial bridging is associated with potentially life-threatening complications including acute MI, tachycardia, syncope, and sudden cardiac death. Myocardial bridges are coronary artery anomalies that are significantly underdiagnosed, and the contemporary literature focuses primarily on symptoms instead of potential genetic causes. Literature has recently emerged proposing the mechanisms behind development, however, research is very limited on identifying genetic and genomic associations. Our systematic review of the literature revealed myocardial bridge development in patients with mutations in MYH7, DPP6, SCN2B, NOTCH1, SLMAP, COMMD10, MACROD2, FBN1, DES, and PRKAG2. Of these genes listed, MYH7, SCN2B, NOTCH1, COMMD10, MACROD2, and FBN1 show the most promise to be candidate genes associated with myocardial bridge development, justifying the further investigation of the mechanisms behind bridge development. Furthermore, recent literature has reported the association of myocardial bridging with specific miRNAs (miR-29b, miR-151-3p, miR-126, miR-503-3p, and miR-645), a tool that may be used for myocardial bridge detection. Additional larger studies into the presence of miRNAs and subsequent myocardial bridge detection are warranted, as these biomarkers may prove to be cost-effective methods for detecting myocardial bridging in both asymptomatic and symptomatic patients. As a definitive molecular basis for myocardial bridge development is lacking, our group has initiated the genomic investigation of a large cohort of myocardial bridge patients within our clinical program and look forward to reporting these data soon. We are hopeful that this review serves to inspire further research into the genomic associations behind myocardial bridge development and spur further interest in this important congenital coronary anomaly.
6. Limitations
This review does not claim a causal role for any gene found to be associated with myocardial bridge development, as this would require the extensive characterization of how each variant affects protein products and eventual phenotype development. Rather, we hope this review will serve as a comprehensive resource of the current genomic associations that may stimulate further hypothesis-driven research into the genomic pathways responsible for myocardial bridge development. We believe that further detailed mechanistic studies regarding the candidate genes of interest will help to better define the role of each mutation and their subsequent phenotypic effects. Further, though we used significant publicly available publication databases including Pubmed, CINAHL, EMBASE, and Cochrane, we acknowledge that there may be other relevant papers that were not found in these selected databases.
Author Contributions
P.M. (Peyton Moore), P.M. (Paul Murdock) and A.R. all contributed equally to the authorship of this entire manuscript. M.S. conceived the hypothesis, supervised manuscript development and editing, and was responsible for the final version. All authors have read and agreed to the published version of the manuscript.
Funding
Though this work was not specifically funded, the authors acknowledge the generous support of the Potishman Foundation, in Fort Worth, Texas, via restricted grants to support the broad research activity of the Sathyamoorthy Lab. Funding number: Potishman Foundation: 02.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data discussed in this report are publicly available as noted in references. No original data is reported.
Acknowledgments
We would like to express our gratitude to the Consultants in Cardiovascular Medicine and Science—Fort Worth for their invaluable contribution by providing us with the necessary background in myocardial bridging and access to their practice database to support the development of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
Myocardial bridging (MB), right coronary artery (RCA), left anterior descending (LAD), left circumflex (LCX), congenital coronary vascular anomalies (CCVAs), computed tomography (CT), hypertrophic cardiomyopathy (HCM), restrictive cardiomyopathy (RCM), sudden cardiac death (SCD), variant of unknown significance (VUS), small interfering RNA (siRNA), array-comparative genomic hybridization (array-CGH), copy number variation (CNV), cullin-RING E3 ubiquitin ligase complexes (CRL), nuclear factor kappa-light-chain-enhancer of activated B cells (NFK-ß), congenital heart disorders (CHD), Wolff-Parkinson-White (WPW), microRNA (miRNA).
References
- Villa, A.D.M.; Sammut, E.; Nair, A.; Rajani, R.; Bonamini, R.; Chiribiri, A. Coronary artery anomalies overview: The normal and the abnormal. World J. Radiol. 2016, 8, 537–555. [Google Scholar] [CrossRef]
- Sternheim, D.; Power, D.A.; Samtani, R.; Kini, A.; Fuster, V.; Sharma, S. Myocardial Bridging: Diagnosis, Functional Assessment, and Management. J. Am. Coll. Cardiol. 2021, 78, 2196–2212. [Google Scholar] [CrossRef] [PubMed]
- Image Created with biorender.com. Available online: http://Biorender.com (accessed on 13 August 2023).
- Picazo, B.; Perez-Pomares, J.M. Human Genetics of Coronary Artery Anomalies. In Congenital Heart Diseases: The Broken Heart; Springer: Vienna, Austria, 2016; pp. 535–539. [Google Scholar]
- Lee, M.S.; Chen, C.H. Myocardial Bridging: An Up-to-Date Review. J. Invasive Cardiol. 2015, 27, 521–528. [Google Scholar] [PubMed]
- With Permission, Research Database—Consultants in Cardiovascular Medicine and Science—Fort Worth—PLLC, Fort Worth, Texas.
- Happach, V.C.; Delk, G.T.; Ganti, L. Myocardial Bridging, the Hidden Risk Factor for Ischemia. Mil. Med. 2022, 187, E1230–E1232. [Google Scholar] [CrossRef] [PubMed]
- Ciliberti, G.; Laborante, R.; Di Francesco, M.; Restivo, A.; Rizzo, G.; Galli, M.; Canonico, F.; Zito, A.; Princi, G.; Vergallo, R.; et al. Comprehensive functional and anatomic assessment of myocardial bridging: Unlocking the Gordian Knot. Front. Cardiovasc. Med. 2022, 9, 970422. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, G.; Mukherjee, D.; Gharacholou, S.M.; Nanjundappa, A.; Lavie, C.J.; Khan, A.A.; Shanmugasundaram, M.; Paul, T.K. An Updated Review on Myocardial Bridging. Cardiovasc. Revascularization Med. 2020, 21, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Greenway, S.C.; Wilson, G.J.; Wilson, J.; George, K.; Kantor, P.F. Sudden Death in an Infant with Angina, Restrictive Cardiomyopathy, and Coronary Artery Bridging An Unusual Phenotype for a beta-Myosin Heavy Chain (MYH7) Sarcomeric Protein Mutation. Circ.-Heart Fail. 2012, 5, E92–E93. [Google Scholar] [CrossRef] [PubMed]
- Grassi, S.; Campuzano, O.; Coll, M.; Brion, M.; Arena, V.; Iglesias, A.; Carracedo, A.; Brugada, R.; Oliva, A. Genetic variants of uncertain significance: How to match scientific rigour and standard of proof in sudden cardiac death? Legal. Med. 2020, 45, 101712. [Google Scholar] [CrossRef]
- Joseph, A.; Hernandez, N.B.; Davies, R.; Tan, W. Managing Myocardial Bridge and Right Ventricular Outflow Tract Obstruction in an Adolescent with Hypertrophic Cardiomyopathy. World J. Pediatr. Congenit. Heart Surg. 2023, 14, 530–532. [Google Scholar] [CrossRef]
- DPP6 Dipeptidyl Peptidase Like 6 [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/1804 (accessed on 15 August 2023).
- Chopra, N.; Knollmann, B.C. Genetics of sudden cardiac death syndromes. Curr. Opin. Cardiol. 2011, 26, 196–203. [Google Scholar] [CrossRef]
- SCN2B Sodium Voltage-Gated Channel Beta Subunit 2 [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/6327 (accessed on 15 August 2023).
- Bao, Y.; Willis, B.C.; Frasier, C.R.; Lopez-Santiago, L.F.; Lin, X.; Ramos-Mondragon, R.; Auerbach, D.S.; Chen, C.; Wang, Z.; Anumonwo, J.; et al. Scn2b Deletion in Mice Results in Ventricular and Atrial Arrhythmias. Circ. Arrhythm. Electrophysiol. 2016, 9, e003923. [Google Scholar] [CrossRef] [PubMed]
- NOTCH1 Notch Receptor 1 [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/4851 (accessed on 20 August 2023).
- Niessen, K.; Karsan, A. Notch signaling in cardiac development. Circ. Res. 2008, 102, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Wang, C.; Xu, Z.; Lin, H.; Wan, X.; Yu, Y.; Adhicary, S.; Zhang, J.Z.; Zhou, Y.; Liu, C.; et al. Impaired Human Cardiac Cell Development due to NOTCH1 Deficiency. Circ. Res. 2023, 132, 187–204. [Google Scholar] [CrossRef]
- Blanco, R.; Gerhardt, H. VEGF and Notch in Tip and Stalk Cell Selection. Cold Spring Harb. Perspect. Med. 2013, 3, a006569. [Google Scholar] [CrossRef]
- Kachanova, O.; Lobov, A.; Malashicheva, A. The Role of the Notch Signaling Pathway in Recovery of Cardiac Function after Myocardial Infarction. Int. J. Mol. Sci. 2022, 23, 12509. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hiroi, Y.; Liao, J.K. Notch Signaling as an Important Mediator of Cardiac Repair and Regeneration after Myocardial Infarction. Trends Cardiovasc. Med. 2010, 20, 228–231. [Google Scholar] [CrossRef]
- Grassi, S.; Vidal, M.C.; Campuzano, O.; Arena, V.; Alfonsetti, A.; Rossi, S.S.; Scarnicci, F.; Iglesias, A.; Brugada, R.; Oliva, A. Sudden Death without a Clear Cause after Comprehensive Investigation: An Example of Forensic Approach to Atypical/Uncertain Findings. Diagnostics 2021, 11, 886. [Google Scholar] [CrossRef]
- Nader, M. The SLMAP/Striatin complex: An emerging regulator of normal and abnormal cardiac excitation-contraction coupling. Eur. J. Pharmacol. 2019, 858, 172491. [Google Scholar] [CrossRef]
- Ishikawa, T.; Sato, A.; Marcou, C.A.; Tester, D.J.; Ackerman, M.J.; Crotti, L.; Schwartz, P.J.; On, Y.K.; Park, J.E.; Nakamura, K.; et al. A Novel Disease Gene for Brugada Syndrome Sarcolemmal Membrane-Associated Protein Gene Mutations Impair Intracellular Trafficking of hNav1.5. Circ.-Arrhythmia Electrophysiol. 2012, 5, 1098–1107. [Google Scholar] [CrossRef]
- Brancaccio, M.; Mennitti, C.; Cesaro, A.; Monda, E.; D’Argenio, V.; Casaburi, G.; Mazzaccara, C.; Ranieri, A.; Fimiani, F.; Barretta, F.; et al. Multidisciplinary In-Depth Investigation in a Young Athlete Suffering from Syncope Caused by Myocardial Bridge. Diagnostics 2021, 11, 2144. [Google Scholar] [CrossRef]
- Lombardo, B.; Esposito, D.; Iossa, S.; Vitale, A.; Verdesca, F.; Perrotta, C.; Di Leo, L.; Costa, V.; Pastore, L.; Franze, A. Intragenic Deletion in MACROD2: A Family with Complex Phenotypes Including Microcephaly, Intellectual Disability, Polydactyly, Renal and Pancreatic Malformations. Cytogenet. Genome Res. 2019, 158, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Hu, B.; Feng, L.; Dong, J.T.; Huang, X.S.; Cai, S.J.; Yuan, Y. A Case of Syncope in a Child due to the Large Segment of Myocardial Bridge. Int. Heart J. 2022, 63, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Ergoren, M.C.; Turkgenc, B.; Teralı, K.; Rodoplu, O.; Verstraeten, A.; Van Laer, L.; Mocan, G.; Loeys, B.; Tetik, O.; Temel, S.G. Identification and characterization of a novel FBN1 gene variant in an extended family with variable clinical phenotype of Marfan syndrome. Connect. Tissue Res. 2019, 60, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.X.; Yu, R.; Sheng, Y.; Fan, L.L.; Deng, Y. Case report: Whole-exome sequencing identifies a novel DES mutation (p. E434K) in a Chinese family with cardiomyopathy and sudden cardiac death. Front. Cardiovasc. Med. 2022, 9, 971501. [Google Scholar] [CrossRef]
- Banankhah, P.; Fishbein, G.A.; Dota, A.; Ardehali, R. Cardiac manifestations of PRKAG2 mutation. BMC Med. Genet. 2018, 19, 1. [Google Scholar] [CrossRef]
- Zhong, Y.; Pei, Y.H.; Wang, J.; Chen, J.; Jiang, S.S.; Gong, J.B. MicroRNA expression profile in myocardial bridging patients. Scand. J. Clin. Lab. Investig. 2014, 74, 582–587. [Google Scholar] [CrossRef]
| Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).




{kind=link}
{kind=link}
{kind=link}
{kind=link}