
Computational Characterization of Undifferentially Expressed Genes with Altered Transcription Regulation in Lung Cancer
 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
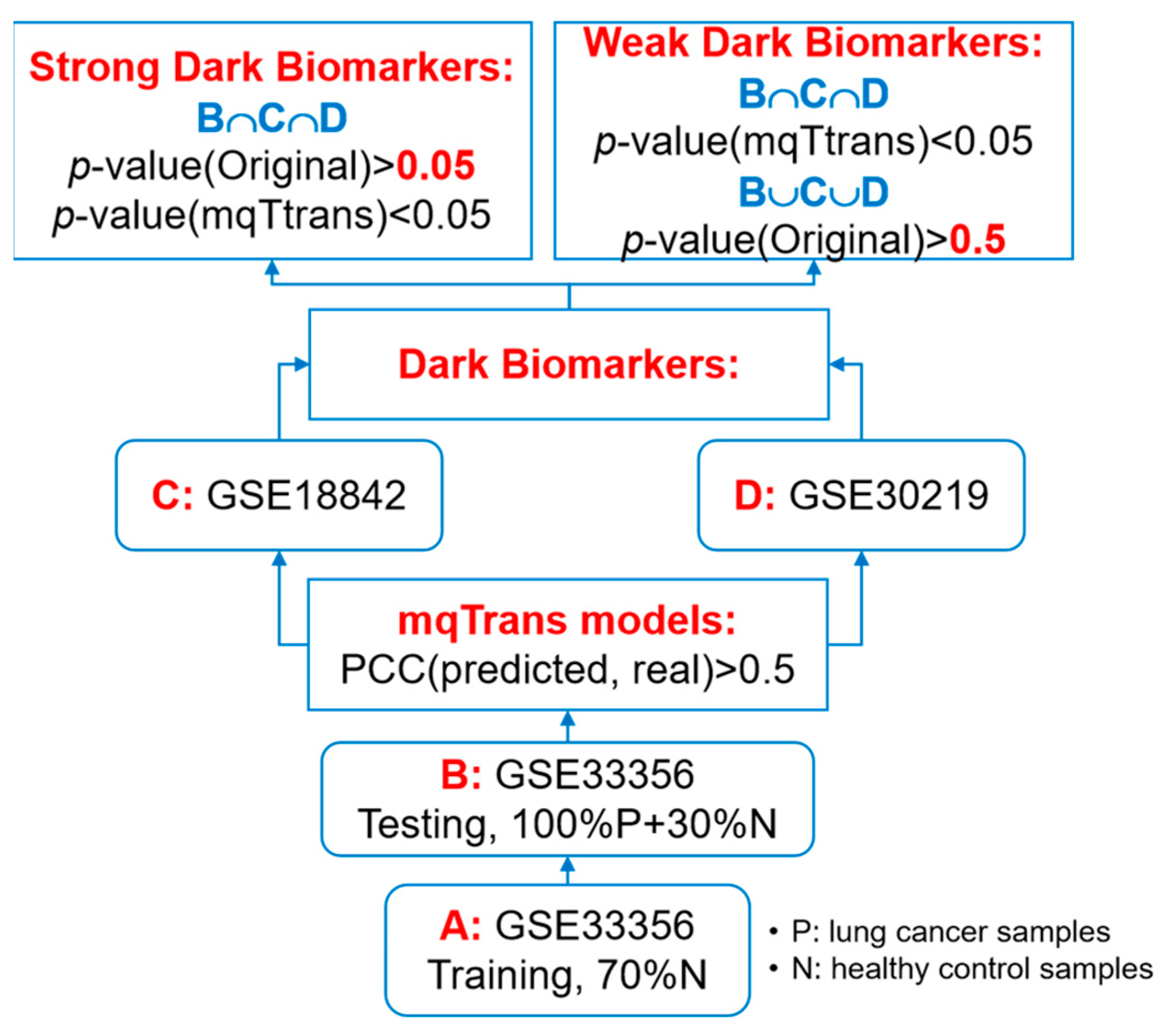
2.1. Summary of the Datasets
2.2. Expression Prediction Using Upstream TFs
2.3. Calculation of the mqTrans Features
2.4. Experimental Design
3. Results and Discussion
3.1. Data Preprocessing
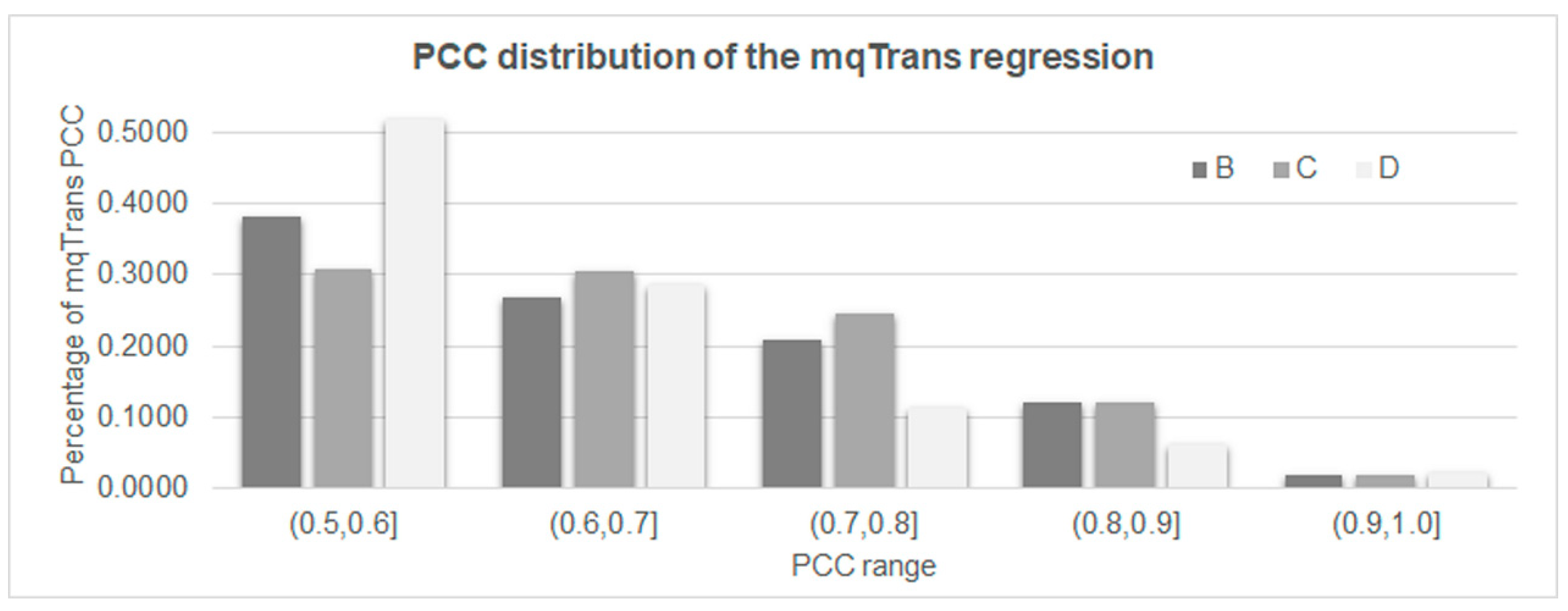
3.2. The Quantitative Transcription Regulatory Models
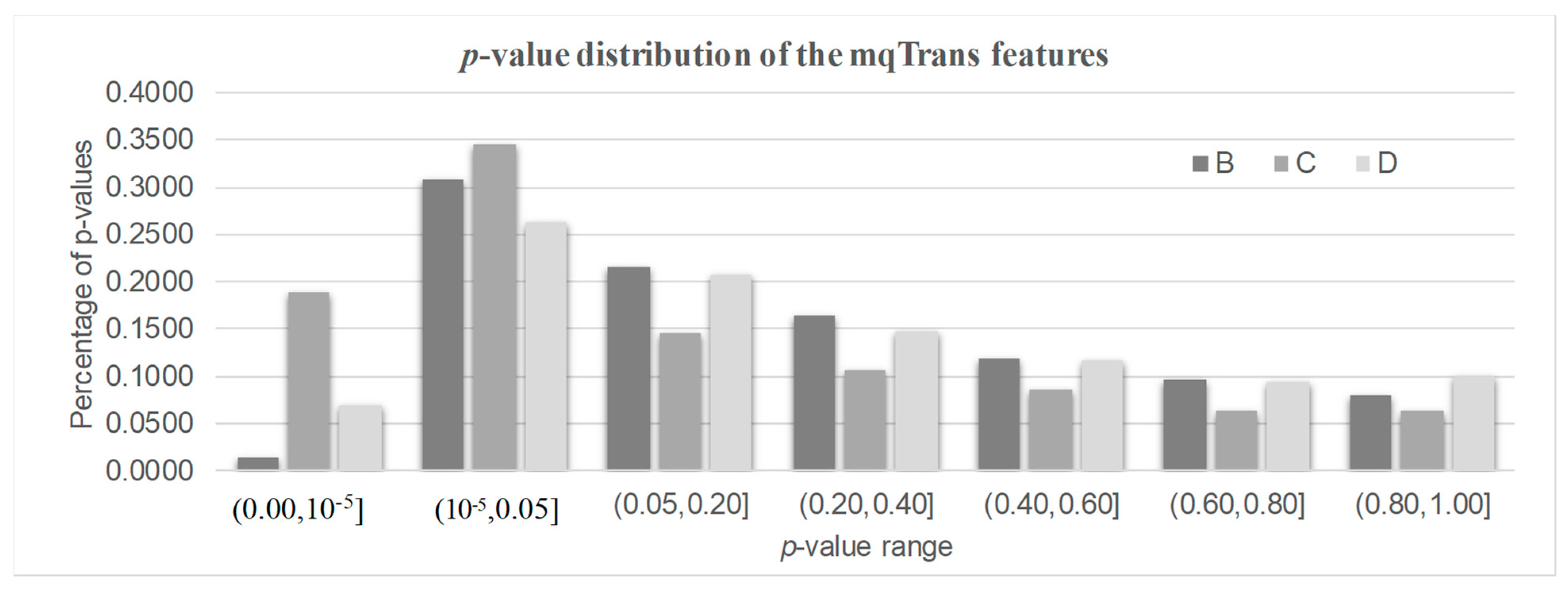
3.3. Differential Transcription Regulation Analysis
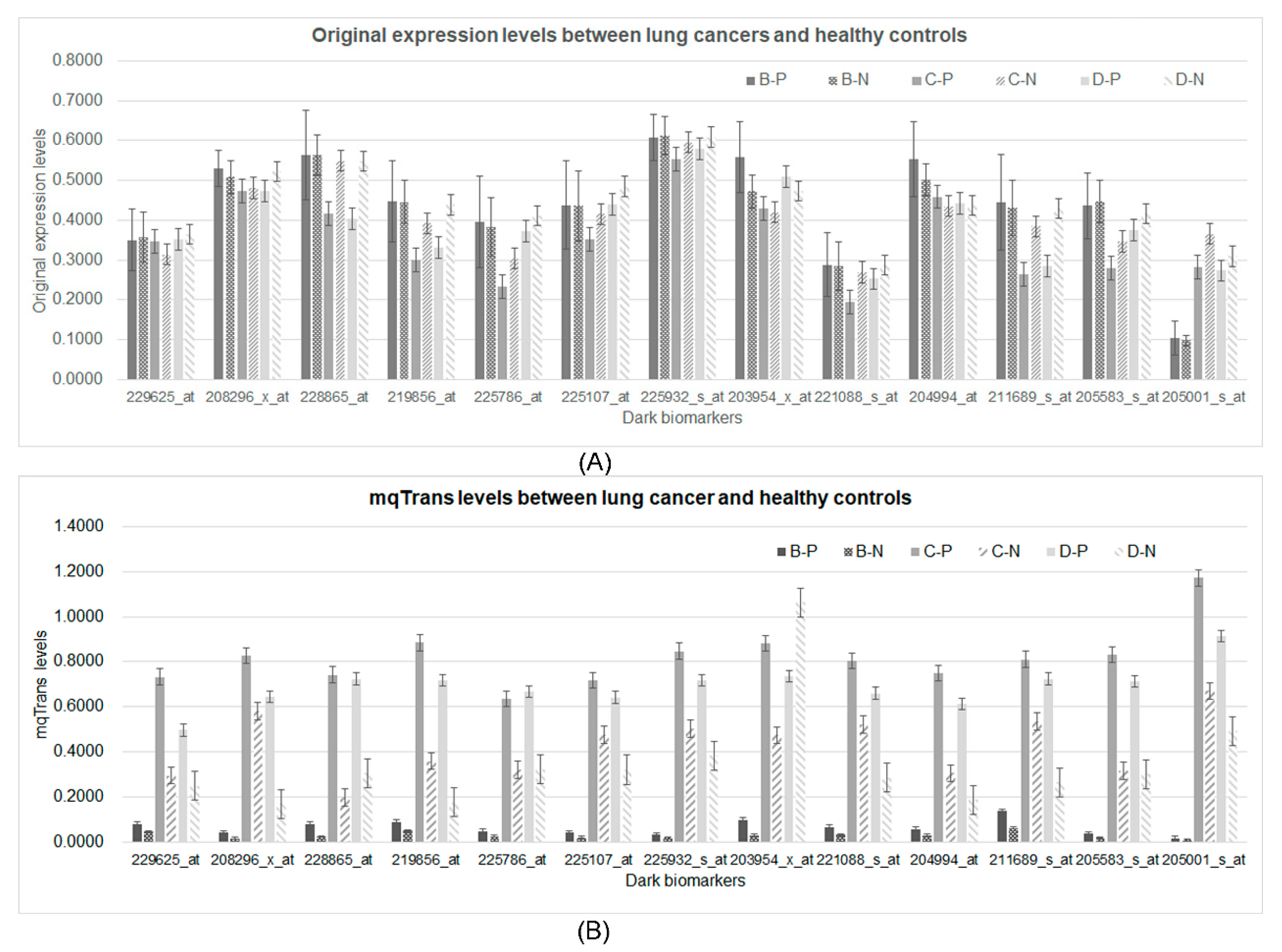
3.4. DeTouR Features Ignored by a Conventional Differential Analysis
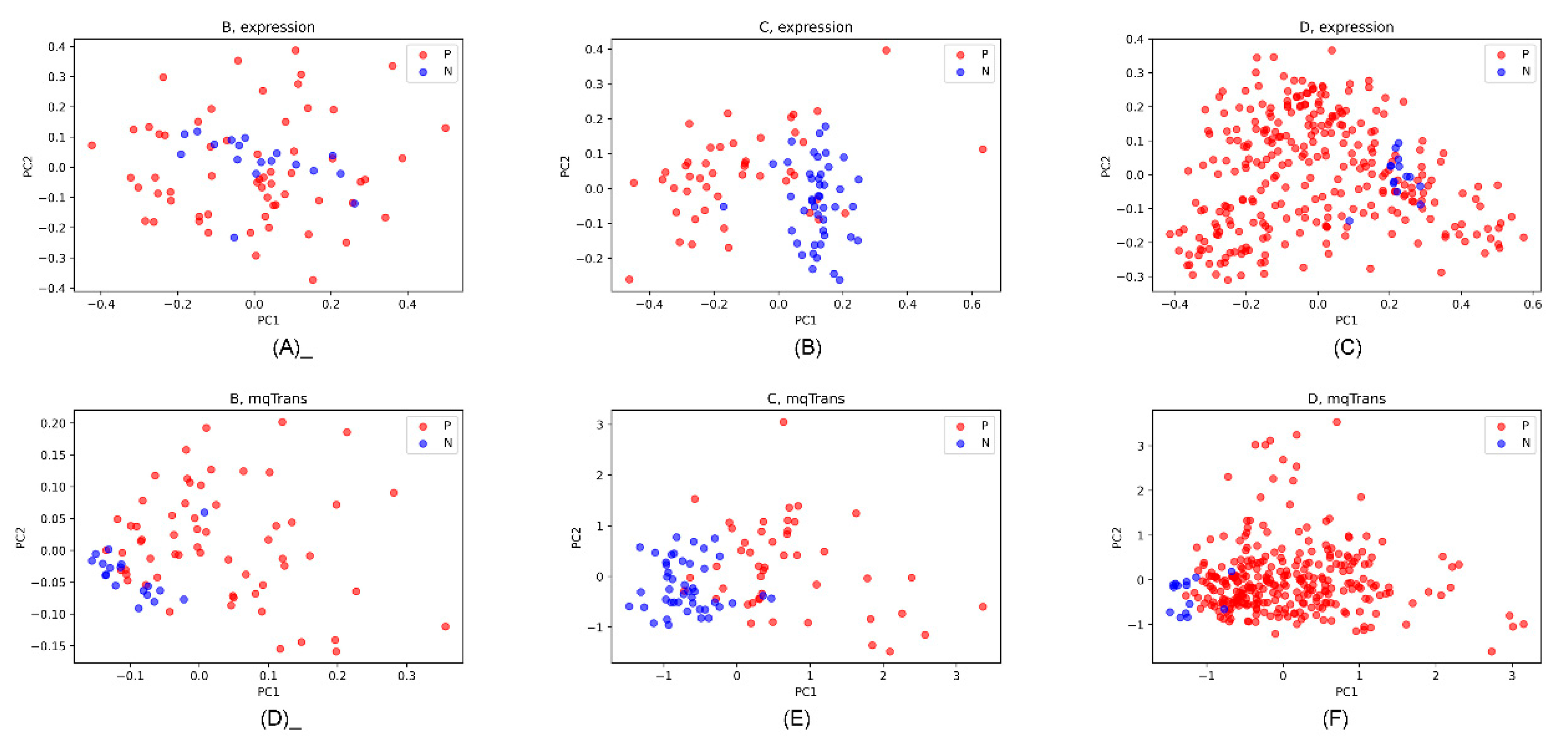
3.5. Differential Patterns in the Two Levels
3.6. Validation of the Dark Biomarkers on an Independent Dataset
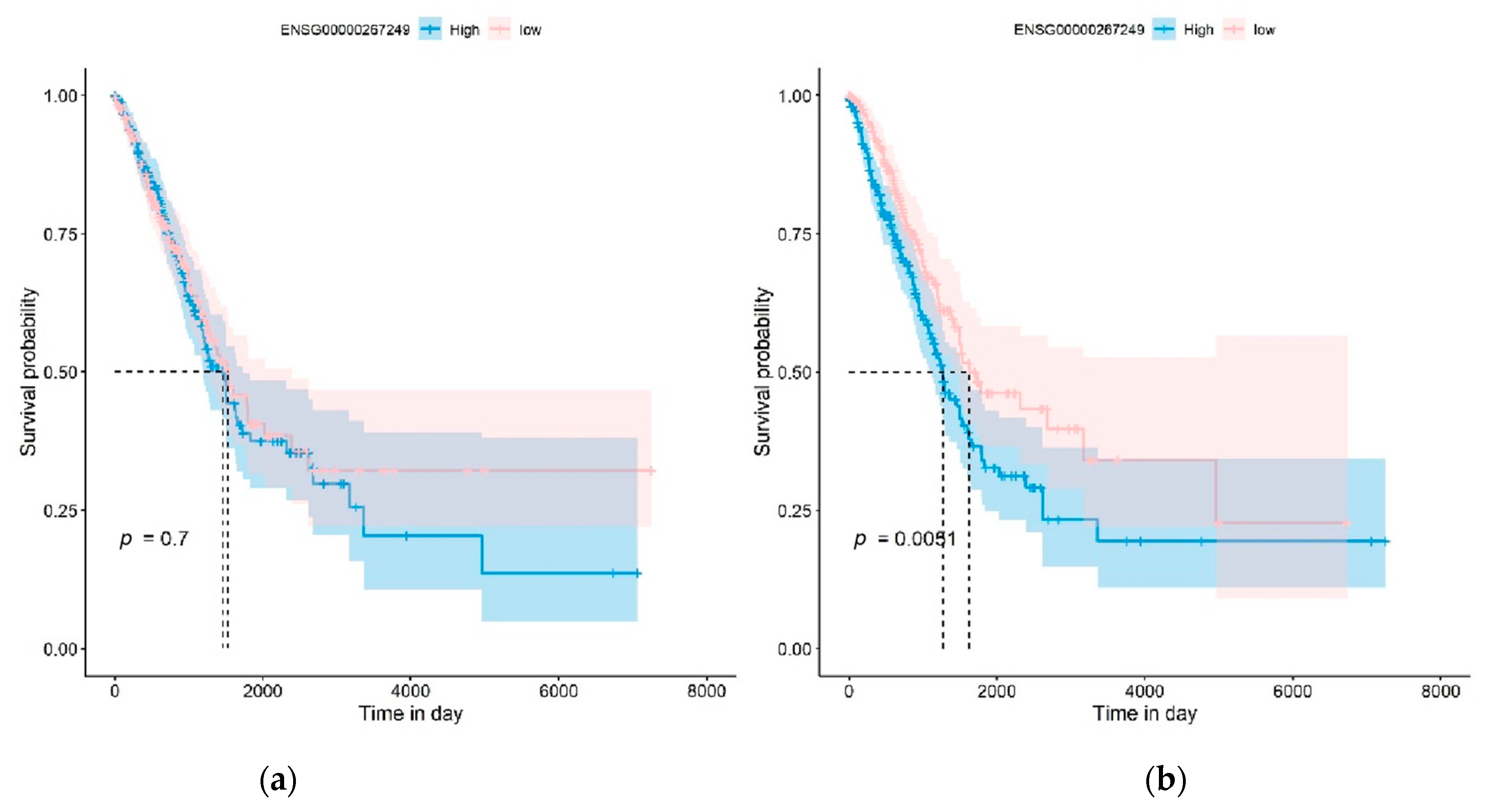
3.7. Biological Observation of the Strong Dark Biomarker GBP5
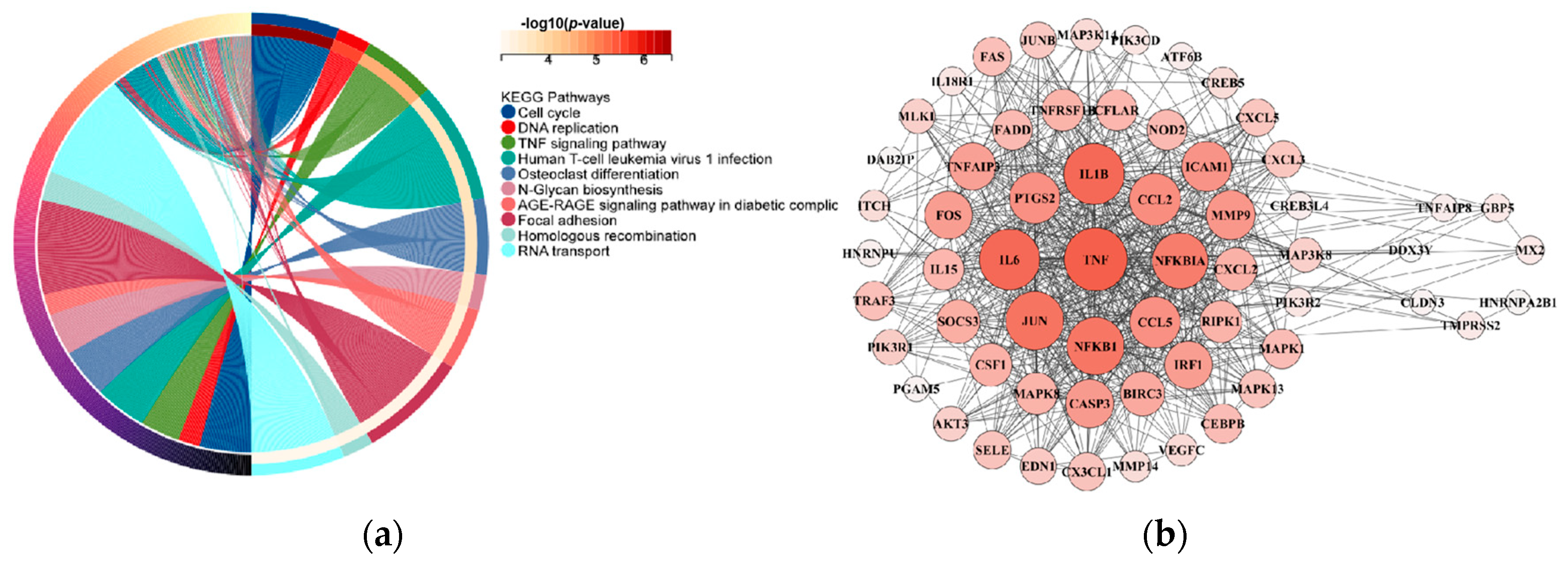
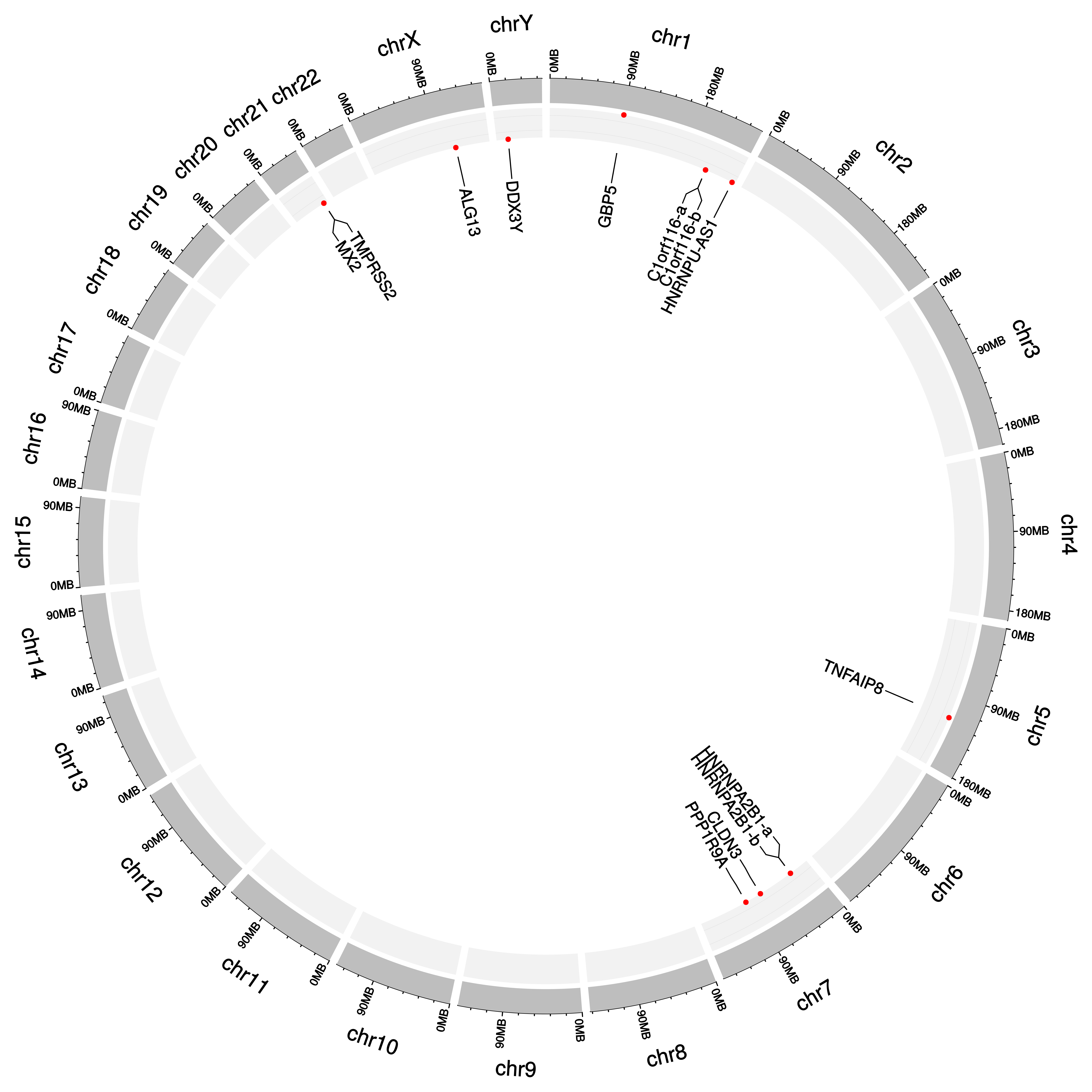
3.8. RNA-Seq Dark Biomarkers of Late-Stage LUAD and LUSC
3.9. Overlapping lncRNAs Could Be a Disturbing Factor
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alberg, A.J.; Samet, J.M. Epidemiology of lung cancer. Chest 2008, 3, 592. [Google Scholar]
- Ren, Y.; Zhao, S.; Jiang, D.; Xin, F.; Zhou, F. Proteomic biomarkers for lung cancer progression. Biomark. Med. 2018, 12, 205. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Tan, D.; Pas, T.D.; Solomon, B.J.; Ahmad, A.; Lazzari, C.; Marinis, F.D.; Spitaleri, G.; Schultz, K.; Friboulet, L. Progression-Free and Overall Survival in ALK-Positive NSCLC Patients Treated with Sequential Crizotinib and Ceritinib. Clin. Cancer Res. 2016, 21, 2745–2752. [Google Scholar] [CrossRef] [PubMed]
- Whang-Peng, J.; Bunn, P.; Kao-Shan, C.S.; Lee, E.C.; Minna, J.D. A nonrandom chromosomal abnormality, del 3p(14-23), in human small cell lung cancer (SCLC). Cancer Genet. Cytogenet. 1982, 6, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Molinier, O.; Goupil, F.; Debieuvre, D.; Auliac, J.B.; Jeandeau, S.; Lacroix, S.; Martin, F.; Grivaux, M. Five-year survival and prognostic factors according to histology in 6101 non-small-cell lung cancer patients. Respir. Med. Res. 2020, 77, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, H.; Zhang, C.; Zhang, C.; Wang, H. Integrative analysis of genomic alteration, immune cells infiltration and prognosis of lung squamous cell carcinoma (LUSC) to identify smoking-related biomarkers. Int. Immunopharmacol. 2020, 89, 107053. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liu, X.; Jie, D.; Wang, X.J.; Xia, L. Differentiated regulation of immune-response related genes between LUAD and LUSC subtypes of lung cancers. Oncotarget 2017, 8, 133–144. [Google Scholar] [CrossRef]
- Gyoba, J.; Shan, S.; Roa, W.; Bédard, E.L.R. Diagnosing Lung Cancers through Examination of Micro-RNA Biomarkers in Blood, Plasma, Serum and Sputum: A Review and Summary of Current Literature. Int. J. Mol. Sci. 2016, 17, 494. [Google Scholar] [CrossRef]
- Syeda, Z.A.; Langden, S.; Munkhzul, C.; Lee, M.; Song, S.J. Regulatory Mechanism of MicroRNA Expression in Cancer. Int. J. Mol. Sci. 2020, 21, 1723. [Google Scholar] [CrossRef]
- Berman, C.G.; Clark, R.A. Diagnostic imaging in cancer. Prim. Care 1992, 19, 677–713. [Google Scholar] [CrossRef]
- Oleksowicz, L.; Morris, J.C.; Phelps, R.G.; Bruckner, H.W. Pulmonary carcinoid presenting as multiple subcutaneous nodules. Tumori 1990, 76, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.; Landau, J.; Ebner, L.; Bütikofer, Y.; Leidolt, L.; Brela, B.; May, M.; Heverhagen, J.; Christe, A. Performance of ultralow-dose CT with iterative reconstruction in lung cancer screening: Limiting radiation exposure to the equivalent of conventional chest X-ray imaging. Eur. Radiol. 2016, 26, 3643–3652. [Google Scholar] [CrossRef] [PubMed]
- Bashir, A.; Gray, M.L.; Hartke, J.; Burstein, D. Nondestructive imaging of human cartilage glycosaminoglycan concentration by MRI. Magn. Reson. Med. 2015, 41, 857–865. [Google Scholar] [CrossRef]
- Pelosi, G.; Sonzogni, A.; Veronesi, G.; Camilli, E.D.; Maisonneuve, P.; Spaggiari, L.; Manzotti, M.; Masullo, M.; Taliento, G.; Fumagalli, C. Pathologic and molecular features of screening low-dose computed tomography (LDCT)-detected lung cancer: A baseline and 2-year repeat study. Lung Cancer 2008, 62, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Morigi, J.J.; Stricker, P.D.; Leeuwen, P.V.; Tang, R.; Ho, B.; Nguyen, Q.; Hruby, G.; Fogarty, G.; Jagavkar, R.; Kneebone, A. Prospective Comparison of 18F-Fluoromethylcholine Versus 68Ga-PSMA PET/CT in Prostate Cancer Patients Who Have Rising PSA after Curative Treatment and Are Being Considered for Targeted Therapy. J. Nucl. Med. 2015, 56, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.; Kirschner, M. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 39, 1005–1010. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef]
- Kulesza, D.W.; Carré, T.; Chouaib, S.; Kaminska, B. Silencing of the transcription factor STAT3 sensitizes lung cancer cells to DNA damaging drugs, but not to TNFα- and NK cytotoxicity. Exp. Cell Res. 2013, 319, 506–516. [Google Scholar] [CrossRef]
- Zheng, W.P.; Flavell, R.A. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. J. Immunol. 2016, 196, 4426–4435. [Google Scholar] [CrossRef]
- Ahmed, H.A.; Nada, O. E2F3 transcription factor: A promising biomarker in lung cancer. Cancer Biomark. 2017, 19, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Song, H.; Wang, C.; Zheng, J.; Zhou, F. Detection and Independent Validation of Model-Based Quantitative Transcriptional Regulation Relationships Altered in Lung Cancers. Front. Bioeng. Biotechnol. 2020, 8, 582. [Google Scholar] [CrossRef] [PubMed]
- Xin, R.; Feng, X.; Zhang, H.; Wang, Y.; Duan, M.; Xie, T.; Dong, L.; Yu, Q.; Huang, L.; Zhou, F. Seven non-differentially expressed ‘dark biomarkers’ show transcriptional dysregulation in chronic lymphocytic leukemia. Pers. Med. 2023, 20, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Edgar, R. Mining microarray data at NCBI’s Gene Expression Omnibus (GEO)*. Methods Mol. Biol. 2006, 338, 175–190. [Google Scholar] [PubMed]
- Dinalankara, W.; Bravo, H.C. Gene Expression Signatures Based on Variability can Robustly Predict Tumor Progression and Prognosis. Cancer Inform. 2015, 14, 71. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Miao, Y.R.; Jia, L.H.; Yu, Q.Y.; Zhang, Q.; Guo, A.Y. AnimalTFDB 3.0: A comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 2019, 47, D33–D38. [Google Scholar] [CrossRef] [PubMed]
- Preacher, K.J.; Curran, P.J.; Bauer, D.J. Computational Tools for Probing Interactions in Multiple Linear Regression, Multilevel Modeling, and Latent Curve Analysis. J. Educ. Behav. Stat. 2006, 31, 437–448. [Google Scholar] [CrossRef]
- Deguines, N.; Brashares, J.S.; Prugh, L.R. Precipitation alters interactions in a grassland ecological community. J. Anim. Ecol. 2017, 86, 262–272. [Google Scholar] [CrossRef]
- Duan, M.; Liu, Y.; Zhao, D.; Li, H.; Zhang, G.; Liu, H.; Wang, Y.; Fan, Y.; Huang, L.; Zhou, F. Gender-specific dysregulations of nondifferentially expressed biomarkers of metastatic colon cancer. Comput. Biol. Chem. 2023, 104, 107858. [Google Scholar] [CrossRef]
- Duan, M.; Zhang, L.; Wang, Y.; Fan, Y.; Liu, S.; Yu, Q.; Huang, L.; Zhou, F. Computational pan-cancer characterization of model-based quantitative transcription regulations dysregulated in regional lymph node metastasis. Comput. Biol. Med. 2021, 135, 104571. [Google Scholar] [CrossRef]
- Diehr, P.; Martin, D.C.; Koepsell, T.; Cheadle, A. Breaking the matches in a paired t-test for community interventions when the number of pairs is small. Stat. Med. 2010, 14, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Clement, E. Using Normalized Bayesian Information Criterion (Bic) to Improve Box-Jenkins Model Building. Am. J. Math. Stat. 2014, 4, 214–221. [Google Scholar] [CrossRef]
- Zhang, L.Q.; Li, Q.Z.; Su, W.X.; Jin, W. Predicting gene expression level by the transcription factor binding signals in human embryonic stem cells. Biosystems 2016, 150, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Beer, M.A.; Tavazoie, S. Predicting gene expression from sequence. Cell 2004, 117, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.Q.; Fornes, O.; Wasserman, W.W. Gene expression models based on transcription factor binding events confer insight into functional cis-regulatory variants. Bioinformatics 2019, 35, 2610–2617. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.R.; Reshef, Y.A.; Bileschi, M.; Belanger, D.; McLean, C.Y.; Snoek, J. Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res. 2018, 28, 739–750. [Google Scholar] [CrossRef]
- Zhang, D.; Xia, J. Somatic synonymous mutations in regulatory elements contribute to the genetic aetiology of melanoma. BMC Med. Genom. 2020, 13, 43. [Google Scholar] [CrossRef]
- Kim, M.; Haney, J.R.; Zhang, P.; Hernandez, L.M.; Wang, L.K.; Perez-Cano, L.; Loohuis, L.M.O.; de la Torre-Ubieta, L.; Gandal, M.J. Brain gene co-expression networks link complement signaling with convergent synaptic pathology in schizophrenia. Nat. Neurosci. 2021, 24, 799–809. [Google Scholar] [CrossRef]
- Tran, S.S.; Zhou, Q.; Xiao, X. Statistical inference of differential RNA-editing sites from RNA-sequencing data by hierarchical modeling. Bioinformatics 2020, 36, 2796–2804. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Kramer, E.D.; Tzetzo, S.L.; Colligan, S.H.; Hensen, M.L.; Brackett, C.M.; Clausen, B.E.; Taketo, M.M.; Abrams, S.I. β-Catenin signaling in alveolar macrophages enhances lung metastasis through a TNF-dependent mechanism. JCI Insight 2023, 8, e160978. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Zhang, H.; Meng, H.; Deng, Z.; Gu, T.; Luo, P.; Zhang, J. TNF-α Pathway Alternation Predicts Survival of Immune Checkpoint Inhibitors in Non-Small Cell Lung Cancer. Front. Immunol. 2021, 12, 667875. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Luo, J.; Ai, N.; Kim, R.; Ahn, L.; Biswas, A.; Coker, C.; Ma, W.; Wong, P.; Buonocore, D.J.; et al. Phase I trial of the TNF-α inhibitor certolizumab plus chemotherapy in stage IV lung adenocarcinomas. Nat. Commun. 2022, 13, 6095. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Ouyang, Y.; Yao, W. shinyCircos: An R/Shiny application for interactive creation of Circos plot. Bioinformatics 2018, 34, 1229–1231. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Shariati-Rad, M.; Hasani, M. Principle component analysis (PCA) and second-order global hard-modelling for the complete resolution of transition metal ions complex formation with 1,10-phenantroline. Anal. Chim. Acta 2009, 648, 60–70. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Hizukuri, Y.; Yamashiro, K.; Makita, N.; Ohnishi, K.; Takeya, M.; Komohara, Y.; Hayashi, Y. Guanylate-binding protein 5 is a marker of interferon-γ-induced classically activated macrophages. Clin. Transl. Immunol. 2016, 5, e111. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Lai, F.; Wang, Y.; Sutter, K.; Dittmer, U.; Ye, J.; Zai, W.; Liu, M.; Shen, F.; et al. Functional Comparison of Interferon-α Subtypes Reveals Potent Hepatitis B Virus Suppression by a Concerted Action of Interferon-α and Interferon-γ Signaling. Hepatology 2021, 73, 486–502. [Google Scholar] [CrossRef]
- Sweeney, T.E.; Braviak, L.; Tato, C.M.; Khatri, P. Genome-wide expression for diagnosis of pulmonary tuberculosis: A multicohort analysis. Lancet Respir. Med. 2016, 4, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Yamakita, I.; Mimae, T.; Tsutani, Y.; Miyata, Y.; Ito, A.; Okada, M. Guanylate binding protein 1 (GBP-1) promotes cell motility and invasiveness of lung adenocarcinoma. Biochem. Biophys. Res. Commun. 2019, 518, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Gou, L.; Wei, T.; Zhang, J. GBP1 promotes erlotinib resistance via PGK1activated EMT signaling in nonsmall cell lung cancer. Int. J. Oncol. 2020, 57, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Cao, J.; Liu, L.; Du, Q.; Li, Z.; Zou, D.; Bajic, V.B.; Zhang, Z. LncBook: A curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, D128–D134. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, L.; Jiang, S.; Li, Q.; Feng, C.; Du, Q.; Zou, D.; Xiao, J.; Zhang, Z.; Ma, L. LncExpDB: An expression database of human long non-coding RNAs. Nucleic Acids Res. 2021, 49, D962–D968. [Google Scholar] [CrossRef]
- Zhao, J.; Guo, C.; Ma, Z.; Liu, H.; Yang, C.; Li, S. Identification of a novel gene expression signature associated with overall survival in patients with lung adenocarcinoma: A comprehensive analysis based on TCGA and GEO databases. Lung Cancer 2020, 149, 90–96. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Li, Q.; Wang, R.; Yang, Z.; Li, W.; Yang, J.; Wang, Z.; Bai, H.; Cui, Y.; Tian, Y.; Wu, Z.; et al. Molecular profiling of human non-small cell lung cancer by single-cell RNA-seq. Genome Med. 2022, 14, 87. [Google Scholar] [CrossRef]
- Chandra, V.; Ibrahim, H.; Halliez, C.; Prasad, R.B.; Vecchio, F.; Dwivedi, O.P.; Kvist, J.; Balboa, D.; Saarimäki-Vire, J.; Montaser, H.; et al. The type 1 diabetes gene TYK2 regulates β-cell development and its responses to interferon-α. Nat. Commun. 2022, 13, 6363. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Shao, J.; Song, H.; Wang, J. The YTH Domain Family of N6-Methyladenosine “Readers” in the Diagnosis and Prognosis of Colonic Adenocarcinoma. BioMed Res. Int. 2020, 2020, 9502560. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Li, X.; Chen, S.; Zhang, G.; Li, K.; Wang, Y.; Duan, M.; Zhou, F.; Liu, H. Transcriptional Dysregulations of Seven Non-Differentially Expressed Genes as Biomarkers of Metastatic Colon Cancer. Genes 2023, 14, 1138. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, C.; Jin, L.; Li, C.; Wang, L. The Prognostic Value of m6A RNA Methylation Regulators in Colon Adenocarcinoma. Med. Sci. Monit. 2019, 25, 9435–9445. [Google Scholar] [CrossRef]
- Tanabe, A.; Tanikawa, K.; Tsunetomi, M.; Takai, K.; Ikeda, H.; Konno, J.; Torigoe, T.; Maeda, H.; Kutomi, G.; Okita, K.; et al. RNA helicase YTHDC2 promotes cancer metastasis via the enhancement of the efficiency by which HIF-1α mRNA is translated. Cancer Lett. 2016, 376, 34–42. [Google Scholar] [CrossRef]
- Yoshimura, K.; Suzuki, Y.; Inoue, Y.; Tsuchiya, K.; Karayama, M.; Iwashita, Y.; Kahyo, T.; Kawase, A.; Tanahashi, M.; Ogawa, H.; et al. CD200 and CD200R1 are differentially expressed and have differential prognostic roles in non-small cell lung cancer. Oncoimmunology 2020, 9, 1746554. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Feature | Gene | dbB | PCC-B | dbC | PCC-C | dbD | PCC-D |
|---|---|---|---|---|---|---|---|---|
| Strong | 229625_at | GBP5 | 1 | 0.5057 | 1 | 0.7275 | 1 | 0.8095 |
| Strong | 208296_x_at | TNFAIP8 | 1 | 0.5395 | 1 | 0.6465 | 1 | 0.6792 |
| Weak | 228865_at | C1orf116 | 1 | 0.5920 | 0 | 0.7263 | 0 | 0.5173 |
| Weak | 219856_at | C1orf116 | 1 | 0.5043 | 0 | 0.5716 | 0 | 0.5186 |
| Weak | 225786_at | HNRNPU-AS1 | 1 | 0.8405 | 0 | 0.7996 | 1 | 0.6197 |
| Weak | 225107_at | HNRNPA2B1 | 1 | 0.9013 | 0 | 0.7447 | 0 | 0.7005 |
| Weak | 225932_s_at | HNRNPA2B1 | 1 | 0.7947 | 0 | 0.6915 | 0 | 0.6522 |
| Weak | 203954_x_at | CLDN3 | 0 | 0.5682 | 1 | 0.5222 | 1 | 0.5282 |
| Weak | 221088_s_at | PPP1R9A | 1 | 0.5592 | 0 | 0.5708 | 1 | 0.6123 |
| Weak | 204994_at | MX2 | 0 | 0.6886 | 1 | 0.6834 | 1 | 0.6351 |
| Weak | 211689_s_at | TMPRSS2 | 1 | 0.5350 | 0 | 0.5364 | 0 | 0.5124 |
| Weak | 205583_s_at | ALG13 | 1 | 0.8589 | 0 | 0.7153 | 0 | 0.5303 |
| Weak | 205001_s_at | DDX3Y | 1 | 0.9180 | 0 | 0.7524 | 1 | 0.7513 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xin, R.; Cheng, Q.; Chi, X.; Feng, X.; Zhang, H.; Wang, Y.; Duan, M.; Xie, T.; Song, X.; Yu, Q.; et al. Computational Characterization of Undifferentially Expressed Genes with Altered Transcription Regulation in Lung Cancer. Genes 2023, 14, 2169. https://doi.org/10.3390/genes14122169
Xin R, Cheng Q, Chi X, Feng X, Zhang H, Wang Y, Duan M, Xie T, Song X, Yu Q, et al. Computational Characterization of Undifferentially Expressed Genes with Altered Transcription Regulation in Lung Cancer. Genes. 2023; 14(12):2169. https://doi.org/10.3390/genes14122169
Chicago/Turabian StyleXin, Ruihao, Qian Cheng, Xiaohang Chi, Xin Feng, Hang Zhang, Yueying Wang, Meiyu Duan, Tunyang Xie, Xiaonan Song, Qiong Yu, and et al. 2023. "Computational Characterization of Undifferentially Expressed Genes with Altered Transcription Regulation in Lung Cancer" Genes 14, no. 12: 2169. https://doi.org/10.3390/genes14122169
APA StyleXin, R., Cheng, Q., Chi, X., Feng, X., Zhang, H., Wang, Y., Duan, M., Xie, T., Song, X., Yu, Q., Fan, Y., Huang, L., & Zhou, F. (2023). Computational Characterization of Undifferentially Expressed Genes with Altered Transcription Regulation in Lung Cancer. Genes, 14(12), 2169. https://doi.org/10.3390/genes14122169