
The Expression and Activation of the NF-κB Pathway Correlate with Methotrexate Resistance and Cell Proliferation in Acute Lymphoblastic Leukemia


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Reagents
2.3. Primary ALL Gene Expression and Gene Set Enrichment Analysis (GSEA)
2.4. Cell Lines’ Gene Expression and Single-Sample GSEA (ssGSEA) Analysis
2.5. Cell Viability Assays
2.6. Determination of the Doubling Time
2.7. Transduction with NF-κB Reporter Vector
2.8. Luminescence Assay with Transduced Cells
2.9. Statistical Analysis
3. Results
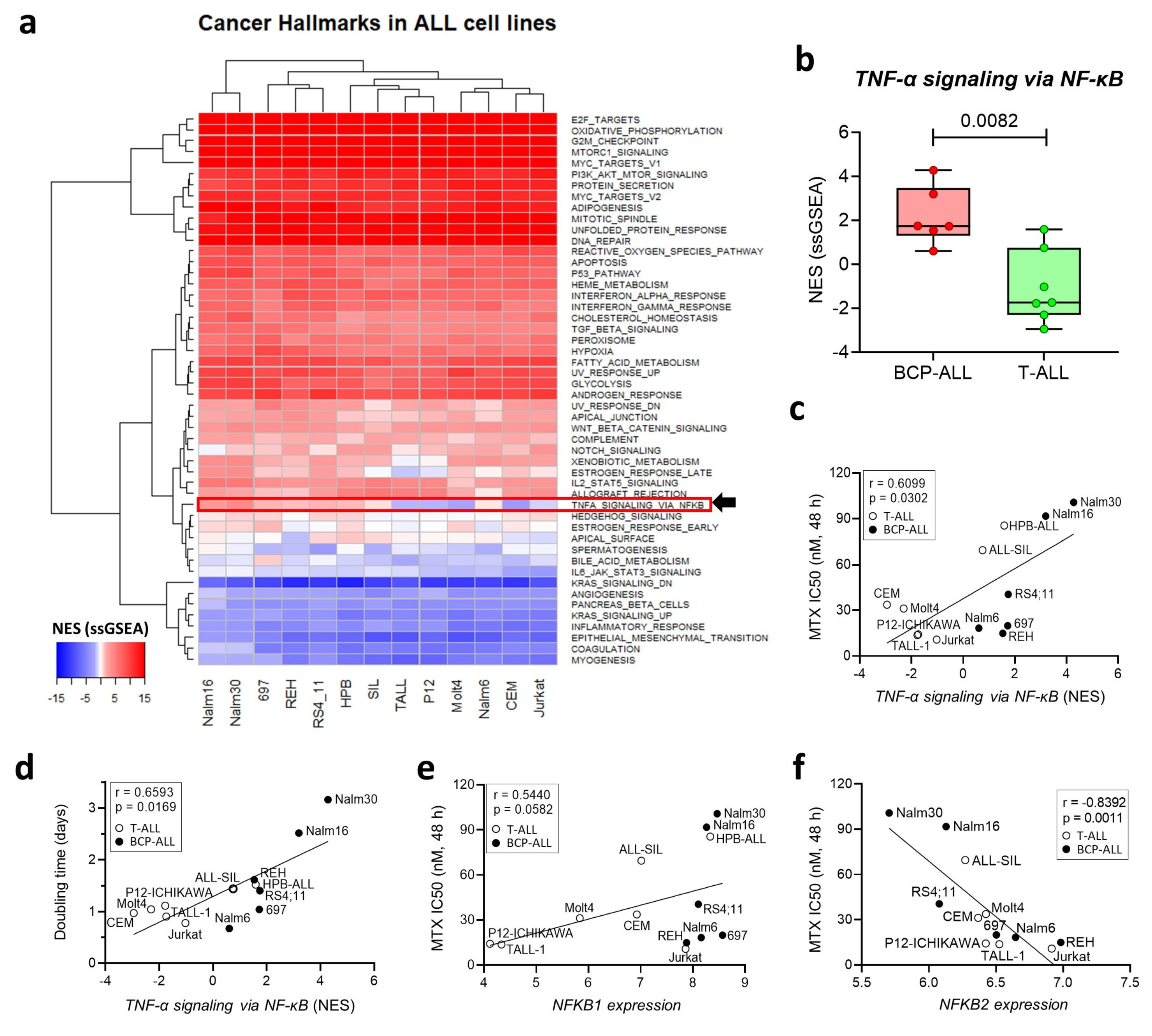
3.1. The NF-κB Signaling Pathway Is Overexpressed in MTX-Resistant Primary ALL
3.2. Expression of the NF-κB Signaling Pathway Is Associated with MTX Resistance in a Panel of ALL Cell Lines
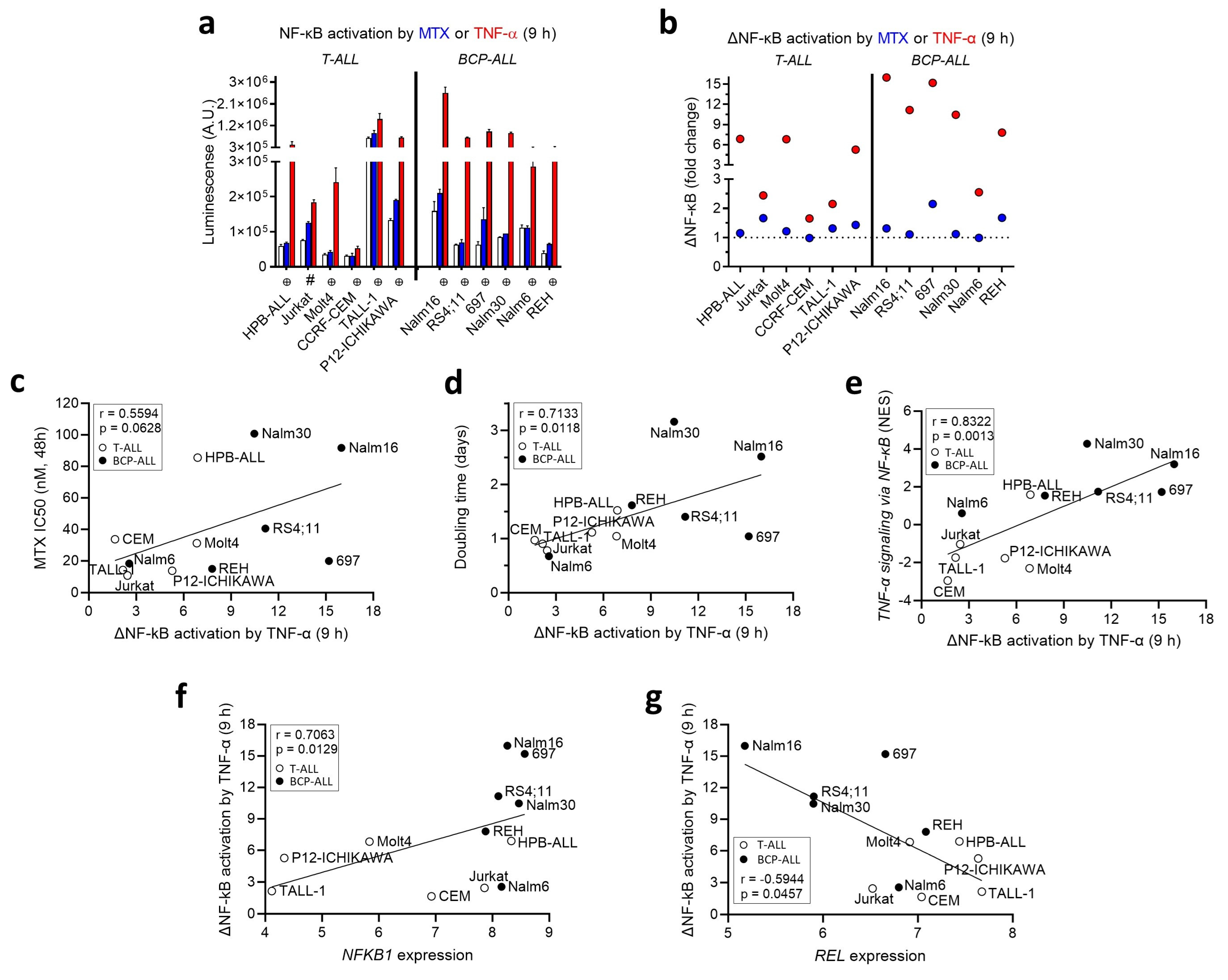
3.3. MTX Modulates NF-κB Activity in a Cell-Line-Dependent Fashion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Spurlock, C.F., 3rd; Tossberg, J.T.; Matlock, B.K.; Olsen, N.J.; Aune, T.M. Methotrexate inhibits NF-kappaB activity via long intergenic (noncoding) RNA-p21 induction. Arthritis Rheumatol. 2014, 66, 2947–2957. [Google Scholar] [CrossRef] [PubMed]
- Spurlock, C.F., 3rd; Gass, H.M.t.; Bryant, C.J.; Wells, B.C.; Olsen, N.J.; Aune, T.M. Methotrexate-mediated inhibition of nuclear factor kappaB activation by distinct pathways in T cells and fibroblast-like synoviocytes. Rheumatology 2015, 54, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Aggarwal, B.B. Methotrexate suppresses NF-kappaB activation through inhibition of IkappaBalpha phosphorylation and degradation. J. Immunol. 2001, 167, 2911–2920. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. The Rel/NF-kappaB signal transduction pathway: Introduction. Oncogene 1999, 18, 6842–6844. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Trzyna, W.C.; McClintock, D.S.; Schumacker, P.T. Role of oxidants in NF-kappa B activation and TNF-αlpha gene transcription induced by hypoxia and endotoxin. J. Immunol. 2000, 165, 1013–1021. [Google Scholar] [CrossRef]
- Qin, H.; Wilson, C.A.; Lee, S.J.; Zhao, X.; Benveniste, E.N. LPS induces CD40 gene expression through the activation of NF-kappaB and STAT-1alpha in macrophages and microglia. Blood 2005, 106, 3114–3122. [Google Scholar] [CrossRef]
- Bosman, M.C.; Schuringa, J.J.; Vellenga, E. Constitutive NF-kappaB activation in AML: Causes and treatment strategies. Crit. Rev. Oncol. Hematol 2016, 98, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Guldenpfennig, C.; Teixeiro, E.; Daniels, M. NF-κB’s contribution to B cell fate decisions. Front Immunol. 2023, 14, 1214095. [Google Scholar] [CrossRef]
- Tsaouli, G.; Barbarulo, A.; Vacca, A.; Screpanti, I.; Felli, M.P. Molecular Mechanisms of Notch Signaling in Lymphoid Cell Lineages Development: NF-kappaB and Beyond. Adv. Exp. Med. Biol. 2020, 1227, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Inda, M.E.; Bagam, P.; Sahoo, M.K.; Osorio, D.; Batra, S. Transcription Factor NF-kappaB: An Update on Intervention Strategies. Arch. Immunol. Ther. Exp 2016, 64, 463–483. [Google Scholar] [CrossRef]
- Li, F.; Zhang, J.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Kumar, A.P.; Ahn, K.S.; Sethi, G. NF-kappaB in cancer therapy. Arch. Toxicol. 2015, 89, 711–731. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, Z.; Zhou, J. Tumor necrosis factor alpha in the onset and progression of leukemia. Exp. Hematol. 2017, 45, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ching, Y.Q.; Chng, W.J. Aberrant nuclear factor-kappa B activity in acute myeloid leukemia: From molecular pathogenesis to therapeutic target. Oncotarget 2015, 6, 5490–5500. [Google Scholar] [CrossRef]
- Iacobucci, I.; Mullighan, C.G. Genetic Basis of Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2017, 35, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Kordes, U.; Krappmann, D.; Heissmeyer, V.; Ludwig, W.D.; Scheidereit, C. Transcription factor NF-kappaB is constitutively activated in acute lymphoblastic leukemia cells. Leukemia 2000, 14, 399–402. [Google Scholar] [CrossRef]
- Dos Santos, N.R.; Ghezzo, M.N.; da Silva, R.C.; Fernandes, M.T. NF-kappaB in T-cell Acute Lymphoblastic Leukemia: Oncogenic Functions in Leukemic and in Microenvironmental Cells. Cancers 2010, 2, 1838–1860. [Google Scholar] [CrossRef]
- Vilimas, T.; Mascarenhas, J.; Palomero, T.; Mandal, M.; Buonamici, S.; Meng, F.; Thompson, B.; Spaulding, C.; Macaroun, S.; Alegre, M.L.; et al. Targeting the NF-kappaB signaling pathway in Notch1-induced T-cell leukemia. Nat. Med. 2007, 13, 70–77. [Google Scholar] [CrossRef]
- Hewamana, S.; Lin, T.T.; Rowntree, C.; Karunanithi, K.; Pratt, G.; Hills, R.; Fegan, C.; Brennan, P.; Pepper, C. Rel a is an independent biomarker of clinical outcome in chronic lymphocytic leukemia. J. Clin. Oncol. 2009, 27, 763–769. [Google Scholar] [CrossRef]
- Hafez, M.; Al-Tonbary, Y.; El-Bayoumi, M.A.; Hatem, N.; Hawas, S.; Mansour, A.; Marzouk, I.; Hafez, M.M.; Yahia, S.; Farahat, N. Markers of apoptosis and proliferation related gene products as predictors of treatment outcome in childhood acute lymphoblastic leukemia. Hematology 2007, 12, 209–218. [Google Scholar] [CrossRef]
- Cleaver, A.L.; Beesley, A.H.; Firth, M.J.; Sturges, N.C.; O’Leary, R.A.; Hunger, S.P.; Baker, D.L.; Kees, U.R. Gene-based outcome prediction in multiple cohorts of pediatric T-cell acute lymphoblastic leukemia: A Children’s Oncology Group study. Mol. Cancer 2010, 9, 105. [Google Scholar] [CrossRef]
- Kamienska, E.; Ociepa, T.; Wysocki, M.; Kurylak, A.; Matysiak, M.; Urasinski, T.; Urasinska, E.; Domagala, W. Activation of NF-κB in leukemic cells in response to initial prednisone therapy in children with acute lymphoblastic leukaemia: Relation to other prognostic factors. Pol. J. Pathol. 2011, 62, 5–11. [Google Scholar] [PubMed]
- Aguayo, A.; Kantarjian, H.; Manshouri, T.; Gidel, C.; Estey, E.; Thomas, D.; Koller, C.; Estrov, Z.; O’Brien, S.; Keating, M.; et al. Angiogenesis in acute and chronic leukemias and myelodysplastic syndromes. Blood 2000, 96, 2240–2245. [Google Scholar] [CrossRef] [PubMed]
- Potapnev, M.P.; Petyovka, N.V.; Belevtsev, M.V.; Savitskiy, V.P.; Migal, N.V. Plasma level of tumor necrosis factor-alpha (TNF-αlpha) correlates with leukocytosis and biological features of leukemic cells, but not treatment response of children with acute lymphoblastic leukemia. Leuk. Lymphoma 2003, 44, 1077–1079. [Google Scholar] [CrossRef] [PubMed]
- Kalmanti, M.; Karamolengou, K.; Dimitriou, H.; Tosca, A.; Vlachonikolis, I.; Peraki, M.; Georgoulias, V.; Kalmantis, T. Serum levels of tumor necrosis factor and soluble interleukin 2 receptor as markers of disease activity and prognosis in childhood leukemia and lymphoma. Int. J. Hematol. 1993, 57, 147–152. [Google Scholar]
- Sorich, M.J.; Pottier, N.; Pei, D.; Yang, W.; Kager, L.; Stocco, G.; Cheng, C.; Panetta, J.C.; Pui, C.H.; Relling, M.V.; et al. In vivo response to methotrexate forecasts outcome of acute lymphoblastic leukemia and has a distinct gene expression profile. PLoS Med. 2008, 5, e83. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Silveira, A.B.; Laranjeira, A.B.A.; Rodrigues, G.O.L.; Leal, R.; Cardoso, B.A.; Barata, J.T.; Yunes, R.A.; Zanchin, N.I.T.; Brandalise, S.R.; Yunes, J.A.; et al. PI3K inhibition synergizes with glucocorticoids but antagonizes with methotrexate in T-cell acute lymphoblastic leukemia. Oncotarget 2015, 6, 13105–13118. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Canevarolo, R.R.; Melo, C.P.S.; Cury, N.M.; Artico, L.L.; Correa, J.R.; Tonhasca Lau, Y.; Mariano, S.S.; Sudalagunta, P.R.; Brandalise, S.R.; Zeri, A.C.M.; et al. Glutathione levels are associated with methotrexate resistance in acute lymphoblastic leukemia cell lines. Front. Oncol. 2022, 12, 1032336. [Google Scholar] [CrossRef]
- Ghashghaeinia, M.; Toulany, M.; Saki, M.; Bobbala, D.; Fehrenbacher, B.; Rupec, R.; Rodemann, H.P.; Ghoreschi, K.; Rocken, M.; Schaller, M.; et al. The NFkB pathway inhibitors Bay 11-7082 and parthenolide induce programmed cell death in anucleated Erythrocytes. Cell Physiol. Biochem. 2011, 27, 45–54. [Google Scholar] [CrossRef]
- Cronstein, B.N.; Aune, T.M. Methotrexate and its mechanisms of action in inflammatory arthritis. Nat. Rev. Rheumatol. 2020, 16, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Mercogliano, M.F.; Bruni, S.; Elizalde, P.V.; Schillaci, R. Tumor Necrosis Factor alpha Blockade: An Opportunity to Tackle Breast Cancer. Front. Oncol. 2020, 10, 584. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canevarolo, R.R.; Cury, N.M.; Yunes, J.A. The Expression and Activation of the NF-κB Pathway Correlate with Methotrexate Resistance and Cell Proliferation in Acute Lymphoblastic Leukemia. Genes 2023, 14, 1880. https://doi.org/10.3390/genes14101880
Canevarolo RR, Cury NM, Yunes JA. The Expression and Activation of the NF-κB Pathway Correlate with Methotrexate Resistance and Cell Proliferation in Acute Lymphoblastic Leukemia. Genes. 2023; 14(10):1880. https://doi.org/10.3390/genes14101880
Chicago/Turabian StyleCanevarolo, Rafael Renatino, Nathalia Moreno Cury, and José Andrés Yunes. 2023. "The Expression and Activation of the NF-κB Pathway Correlate with Methotrexate Resistance and Cell Proliferation in Acute Lymphoblastic Leukemia" Genes 14, no. 10: 1880. https://doi.org/10.3390/genes14101880
APA StyleCanevarolo, R. R., Cury, N. M., & Yunes, J. A. (2023). The Expression and Activation of the NF-κB Pathway Correlate with Methotrexate Resistance and Cell Proliferation in Acute Lymphoblastic Leukemia. Genes, 14(10), 1880. https://doi.org/10.3390/genes14101880