

Statistical Mechanics Metrics in Pairing and Parsing In Silico and Phenotypic Data of a Novel Genetic NFκB1 (c.T638A) Variant
 , ,
, , 
Abstract
:1. Introduction
2. Case Report
3. Materials and Methods
3.1. Ethical Compliance
3.2. Molecular Modeling
3.3. Molecular Dynamics Simulations
4. Results
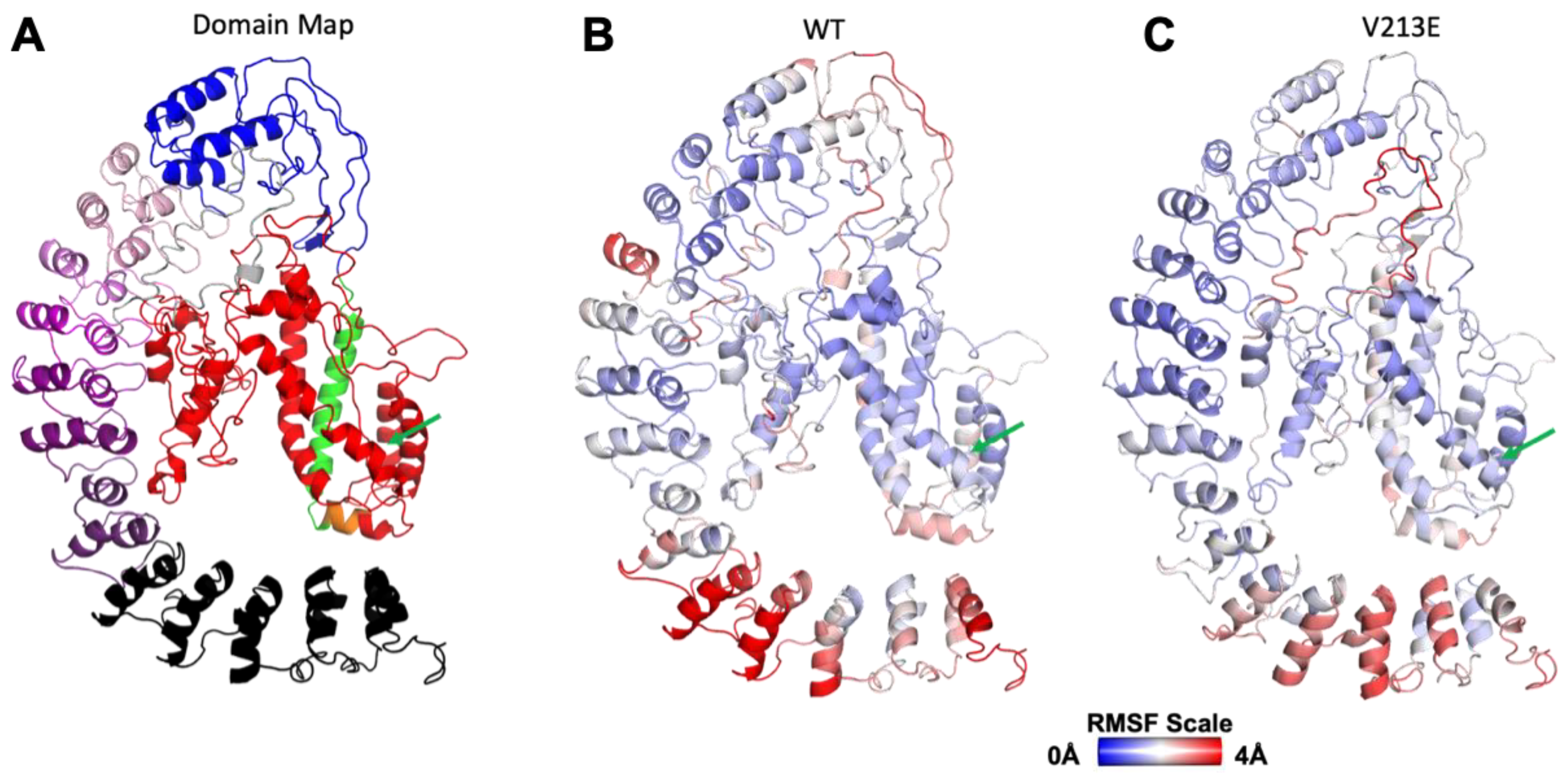
4.1. Gross Structure and Domain Map of Wild Type and Novel Variant V213E NFκB1
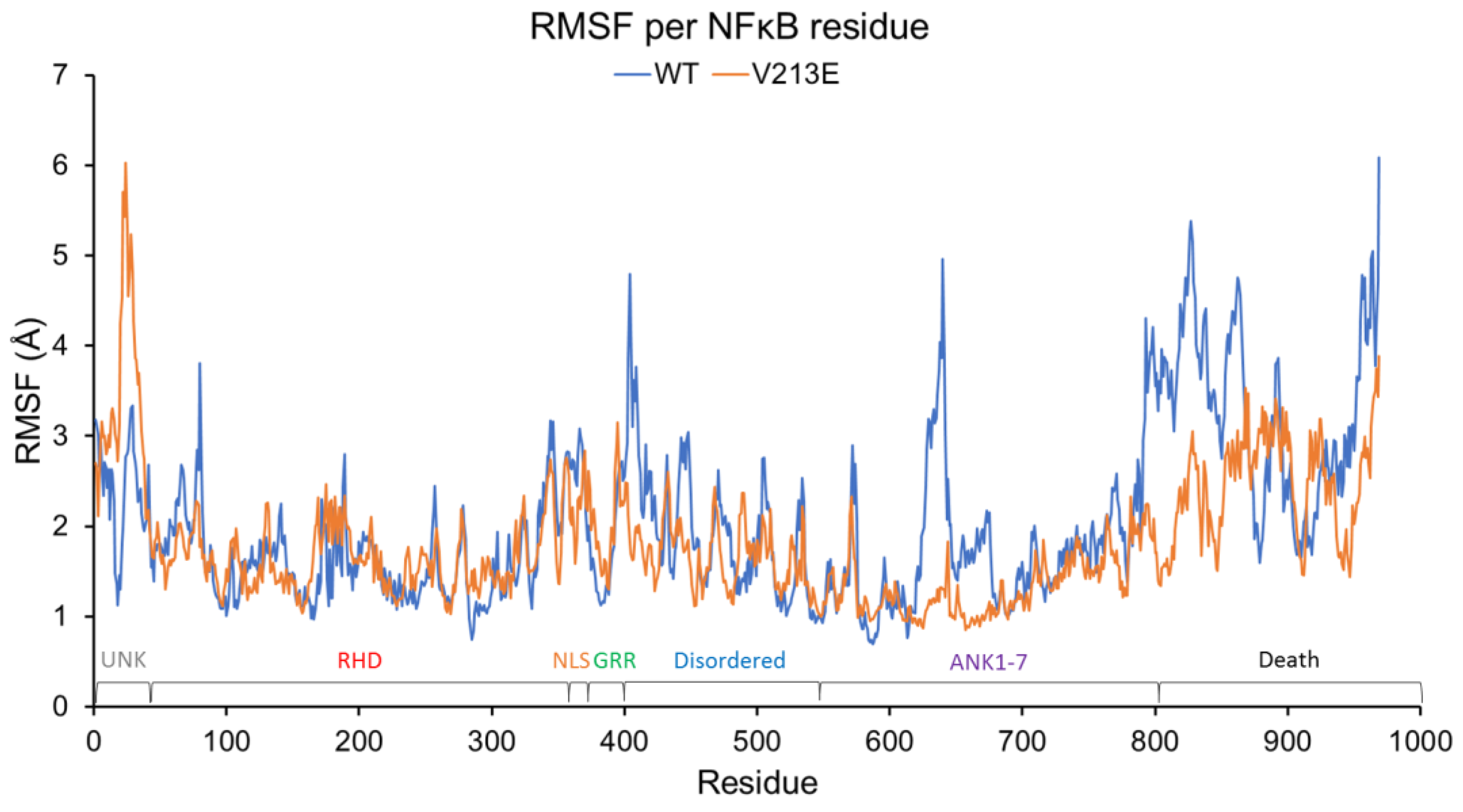
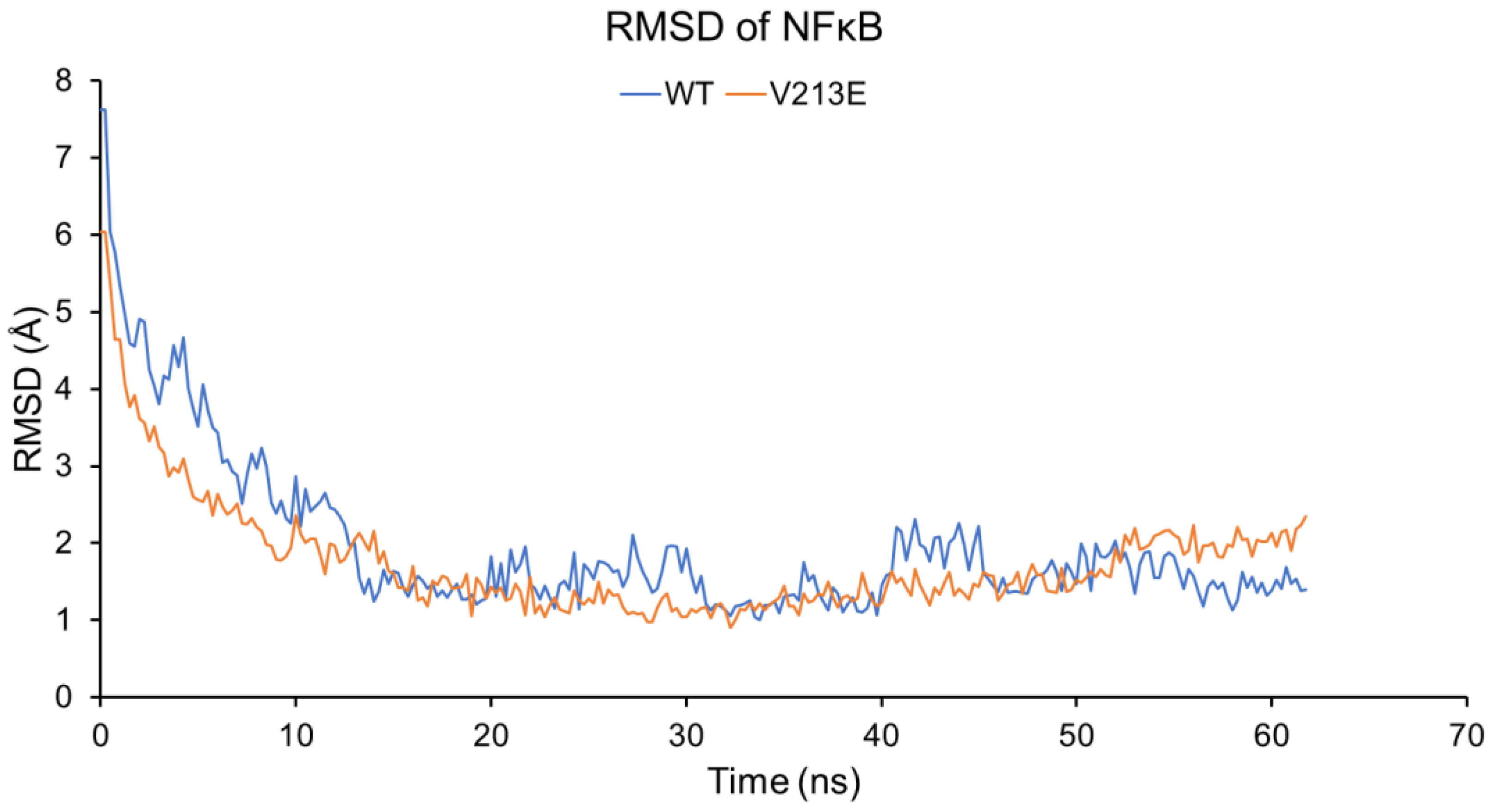
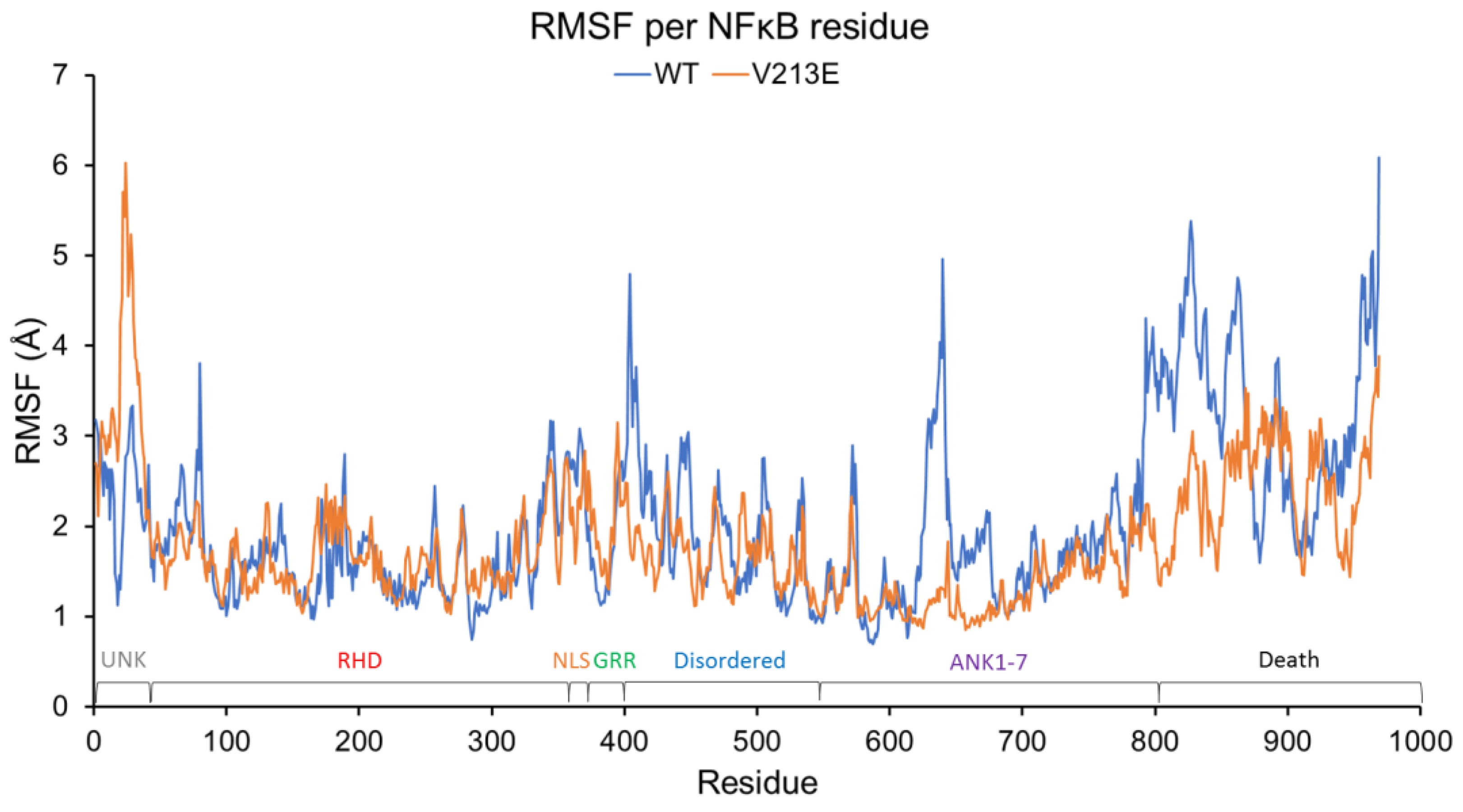
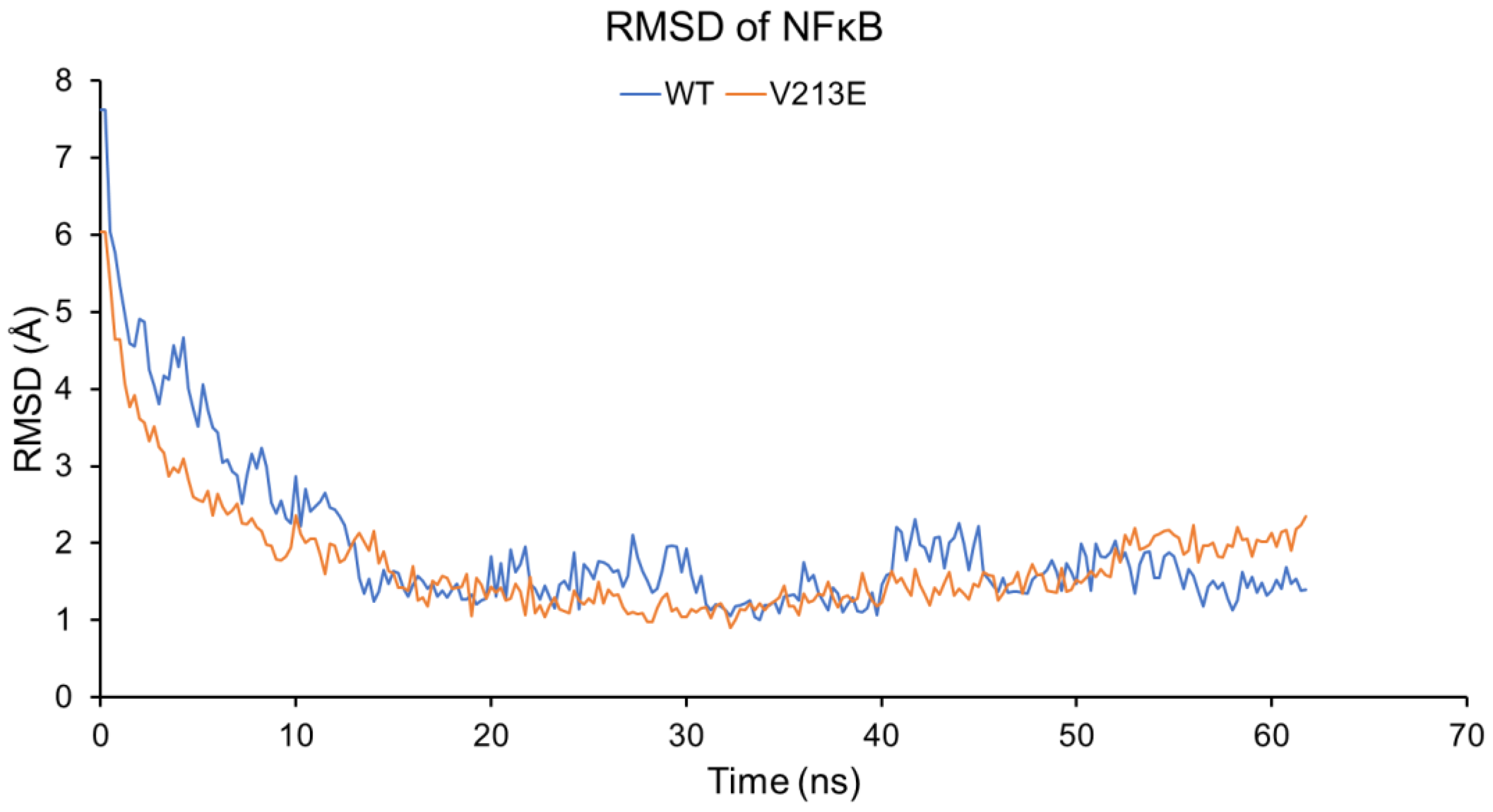
4.2. Molecular Dynamic Simulations via YASARA2 Software
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| “MAEDDPYLGRPEQMFHLDPSLTHTIFNPEVFQPQMALPTADGPYLQILEQPKQRGFRFRYVCE GPSHGGLPGASSEKNKKSYPQVKICNYVGPAKVIVQLVTNGKNIHLHAHSLVGKHCEDGICTVT AGPKDMVVGFANLGILHVTKKKVFETLEARMTEACIRGYNPGLLVHPDLAYLQAEGGGDRQL GDREKELIRQAALQQTKEMDLSVERLMFTAFLPDSTGSFTRRLEPVVSDAIYDSKAPNASNLKIV RMDRTAGCVTGGEEIYLLCDKVQKDDIQIRFYEEEENGGVWEGFGDFSPTDVHRQFAIVFKTPK YKDINITKPASVFVQLRRKSDLETSEPKPFLYYPEIKDKEEVQRKRQKLMPNFSDSFGGGSGAGAG GGGMFGSGGGGGGTGSTGPGYSFPHYGFPTYGGITFHPGTTKSNAGMKHGTMDTESKKDPEG CDKSDDKNTVNLFGKVIETTEQDQEPSEATVGNGEVTLTYATGTKEESAGVQDNLFLEKAMQL AKRHANALFDYAVTGDVKMLLAVQRHLTAVQDENGDSVLHLAIIHLHSQLVRDLLEVTSGLISD DIINMRNDLYQTPLHLAVITKQEDVVEDLLRAGADLSLLDRLGNSVLHLAAKEGHDKVLSILLK HKKAALLLDHPNGDGLNAIHLAMMSNSLPCLLLLVAAGADVNAQEQKSGRTALHLAVEHDN ISLAGCLLLEGDAHVDSTTYDGTTPLHIAAGRGSTRLAALLKAAGADPLVENFEPLYDLDDSWE NAGEDEGVVPGTTPLDMATSWQVFDILNGKPYEPEFTSDDLLAQGDMKQLAEDVKLQLYKLLE IPDPDKNWATLAQKLGLGILNNAFRLSPAPSKTLMDNYEVSGGTVRELVEALRQMGYTEAIEVI QAASSPVKTTSQAHSLPLSPASTRQQIDELRDSDSVCDSGVETSFRKLSFTESLTSGASLLTLNKMP HDYGQEGPLEGKI” |
References
- Hoffmann, A.; Baltimore, D. Circuitry of nuclear factor κB signaling. Immunol. Rev. 2006, 210, 171–186. [Google Scholar] [CrossRef]
- Tieri, P.; Termanini, A.; Bellavista, E.; Salvioli, S.; Capri, M.; Franceschi, C. Charting the NF-κB pathway interactome map. PLoS ONE 2012, 7, e32678. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef] [PubMed]
- Somma, D.; Kok, F.O.; Kerrigan, D.; Wells, C.A.; Carmody, R.J. Defining the Role of Nuclear Factor (NF)-κB p105 Subunit in Human Macrophage by Transcriptomic Analysis of NFKB1 Knockout THP1 Cells. Front. Immunol. 2021, 12, 669906. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Z.; Tang, E.; Fan, Z.; McCauley, L.; Franceschi, R.; Guan, K.; Krebsbach, P.H.; Wang, C.Y. Inhibition of osteoblastic bone formation by nuclear factor-κB. Nat. Med. 2009, 15, 682–689. [Google Scholar] [CrossRef]
- Baldwin, A.S., Jr. The NF-κB and IκB proteins: New discoveries and insights. Annu. Rev. Immunol. 1996, 14, 649–683. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, G.; van Duyne, G.; Ghosh, S.; Sigler, P.B. Structure of NF-κB p50 homodimer bound to a κB site. Nature 1995, 373, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lei, W.T.; Zhang, P.; Rapaport, F.; Seeleuthner, Y.; Lyu, B.; Asano, T.; Rosain, J.; Hammadi, B.; Zhang, Y.; et al. Biochemically deleterious human NFKB1 variants underlie an autosomal dominant form of common variable immunodeficiency. J. Exp. Med. 2021, 218, e20210566. [Google Scholar] [CrossRef]
- Yu, Y.; Wan, Y.; Huang, C. The biological functions of NF-κB1 (p50) and its potential as an anti-cancer target. Curr. Cancer Drug Targets 2009, 9, 566–571. [Google Scholar] [CrossRef]
- Jia, S.; Flores-Saaib, R.D.; Courey, A.J. The Dorsal Rel homology domain plays an active role in transcriptional regulation. Mol. Cell Biol. 2002, 22, 5089–5099. [Google Scholar] [CrossRef]
- Lin, L.; Ghosh, S. A glycine-rich region in NF-κB p105 functions as a processing signal for the generation of the p50 subunit. Mol. Cell Biol. 1996, 16, 2248–2254. [Google Scholar] [CrossRef]
- Hatada, E.N.; Nieters, A.; Wulczyn, F.G.; Naumann, M.; Meyer, R.; Nucifora, G.; McKeithan, T.W.; Scheidereit, C. The ankyrin repeat domains of the NF-κB precursor p105 and the protooncogene bcl-3 act as specific inhibitors of NF-κB DNA binding. Proc. Natl. Acad. Sci. USA 1992, 89, 2489–2493. [Google Scholar] [CrossRef]
- Beinke, S.; Belich, M.P.; Ley, S.C. The death domain of NF-κB1 p105 is essential for signal-induced p105 proteolysis. J. Biol. Chem. 2002, 277, 24162–24168. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Beinke, S.; Ley, S.C. Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem. J. 2004, 382, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Lorenzini, T.; Fliegauf, M.; Klammer, N.; Frede, N.; Proietti, M.; Bulashevska, A.; Camacho-Ordonez, N.; Varjosalo, M.; Kinnunen, M.; de Vries, E.; et al. Characterization of the clinical and immunologic phenotype and management of 157 individuals with 56 distinct heterozygous NFKB1 mutations. J. Allergy Clin. Immunol. 2020, 146, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Tuijnenburg, P.; Lango Allen, H.; Burns, S.O.; Greene, D.; Jansen, M.H.; Staples, E.; Stephens, J.; Carss, K.J.; Biasci, D.; Baxendale, H.; et al. Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J. Allergy Clin. Immunol. 2018, 142, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, F.A.; Barlan, I.; Chapel, H.; Costa-Carvalho, B.T.; Cunningham-Rundles, C.; de la Morena, M.T.; Espinosa-Rosales, F.J.; Hammarstrom, L.; Nonoyama, S.; Quinti, I.; et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. J. Allergy Clin. Immunol. Pract. 2016, 4, 38–59. [Google Scholar] [CrossRef]
- Cunningham-Rundles, C. Common variable immune deficiency: Case studies. Blood 2019, 134, 1787–1795. [Google Scholar] [CrossRef]
- Weifenbach, N.; Schneckenburger, A.A.C.; Lotters, S. Global Distribution of Common Variable Immunodeficiency (CVID) in the Light of the UNDP Human Development Index (HDI): A Preliminary Perspective of a Rare Disease. J. Immunol. Res. 2020, 2020, 8416124. [Google Scholar] [CrossRef] [PubMed]
- Fliegauf, M.; Bryant, V.L.; Frede, N.; Slade, C.; Woon, S.T.; Lehnert, K.; Winzer, S.; Bulashevska, A.; Scerri, T.; Leung, E.; et al. Haploinsufficiency of the NF-κB1 Subunit p50 in Common Variable Immunodeficiency. Am. J. Hum. Genet. 2015, 97, 389–403. [Google Scholar] [CrossRef] [PubMed]
- World Medical, A. World Medical Association Declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA 2013, 310, 2191–2194. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Zidek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- Macklin, S.K.; Bruno, K.A.; Vadlamudi, C.; Helmi, H.; Samreen, A.; Mohammad, A.N.; Hines, S.; Atwal, P.S.; Caulfield, T.R. Examination of Molecular Effects of MYLK Deletion in a Patient with Extensive Aortic, Carotid, and Abdominal Dissections That Underlie the Genetic Dysfunction. Case Rep. Med. 2020, 2020, 5108052. [Google Scholar] [CrossRef]
- Blackburn, P.R.; Sullivan, A.E.; Gerassimou, A.G.; Kleinendorst, L.; Bersten, D.C.; Cooiman, M.; Harris, K.G.; Wierenga, K.J.; Klee, E.W.; van Gerpen, J.A.; et al. Functional Analysis of the SIM1 Variant p.G715V in 2 Patients With Obesity. J. Clin. Endocrinol. Metab. 2020, 105, 355–361. [Google Scholar] [CrossRef]
- Blackburn, P.R.; Carter, J.M.; Oglesbee, D.; Westendorf, J.J.; Neff, B.A.; Stichel, D.; Tsen, D.W.; Gavrilova, R.H.; Wesseling, P.; von Deimling, A.; et al. An activating germline IDH1 variant associated with a tumor entity characterized by unilateral and bilateral chondrosarcoma of the mastoid. HGG Adv. 2020, 1, 100006. [Google Scholar] [CrossRef]
- Richter, J.E., Jr.; Samreen, A.; Vadlamudi, C.; Helmi, H.; Mohammad, A.N.; Wierenga, K.; Hines, S.; Atwal, P.S.; Caulfield, T.R. Genomic Observations of a Rare/Pathogenic SMAD3 Variant in Loeys(-)Dietz Syndrome 3 Confirmed by Protein Informatics and Structural Investigations. Medicina 2019, 55, 137. [Google Scholar] [CrossRef]
- Hines, S.L.; Richter, J.E., Jr.; Mohammad, A.N.; Mahim, J.; Atwal, P.S.; Caulfield, T.R. Protein informatics combined with multiple data sources enriches the clinical characterization of novel TRPV4 variant causing an intermediate skeletal dysplasia. Mol. Genet. Genom. Med. 2019, 7, e566. [Google Scholar] [CrossRef]
- Hines, S.L.; Mohammad, A.N.; Jackson, J.; Macklin, S.; Caulfield, T.R. Integrative data fusion for comprehensive assessment of a novel CHEK2 variant using combined genomics, imaging, and functional-structural assessments via protein informatics. Mol. Omics 2019, 15, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.E., Jr.; Zimmermann, M.T.; Blackburn, P.R.; Mohammad, A.N.; Klee, E.W.; Pollard, L.M.; Macmurdo, C.F.; Atwal, P.S.; Caulfield, T.R. Protein modeling and clinical description of a novel in-frame GLB1 deletion causing GM1 gangliosidosis type II. Mol. Genet. Genom. Med. 2018, 6, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.E.; Robles, H.G.; Mauricio, E.; Mohammad, A.; Atwal, P.S.; Caulfield, T.R. Protein molecular modeling shows residue T599 is critical to wild-type function of POLG and description of a novel variant associated with the SANDO phenotype. Hum. Genome Var. 2018, 5, 18016. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Kohnke, B.; Kutzner, C.; Grubmuller, H. A GPU-Accelerated Fast Multipole Method for GROMACS: Performance and Accuracy. J. Chem. Theory Comput. 2020, 16, 6938–6949. [Google Scholar] [CrossRef]
- Norris, G.A.; Tsai, A.C.; Schneider, K.W.; Wu, Y.H.; Caulfield, T.; Green, A.L. A novel, germline, deactivating CBL variant p.L493F alters domain orientation and is associated with multiple childhood cancers. Cancer Genet. 2021, 254–255, 18–24. [Google Scholar] [CrossRef]
- Liu, C.C.; Murray, M.E.; Li, X.; Zhao, N.; Wang, N.; Heckman, M.G.; Shue, F.; Martens, Y.; Li, Y.; Raulin, A.C.; et al. APOE3-Jacksonville (V236E) variant reduces self-aggregation and risk of dementia. Sci. Transl. Med. 2021, 13, eabc9375. [Google Scholar] [CrossRef]
- Coban, M.A.; Morrison, J.; Maharjan, S.; Hernandez Medina, D.H.; Li, W.; Zhang, Y.S.; Freeman, W.D.; Radisky, E.S.; Le Roch, K.G.; Weisend, C.M.; et al. Attacking COVID-19 Progression Using Multi-Drug Therapy for Synergetic Target Engagement. Biomolecules 2021, 11, 787. [Google Scholar] [CrossRef]
- Richter, J.E., Jr.; Vadlamudi, C.; Macklin, S.K.; Samreen, A.; Helmi, H.; Broderick, D.; Mohammad, A.N.; Hines, S.L.; VanGerpen, J.A.; Atwal, P.S.; et al. Characterization of a Pathogenic Variant in the ABCD1 Gene Through Protein Molecular Modeling. Case Rep. Genet. 2020, 2020, 3256539. [Google Scholar] [CrossRef]
- Land, H.; Humble, M.S. YASARA: A Tool to Obtain Structural Guidance in Biocatalytic Investigations. Methods Mol. Biol. 2018, 1685, 43–67. [Google Scholar] [CrossRef]
- Polak, G.R.E. Note sur la convergence de méthodes de directions conjuguées. In Série Rouge; 1969; Volume 3, pp. 35–43. [Google Scholar]
- Harvey, M.J.; De Fabritiis, G. An Implementation of the Smooth Particle Mesh Ewald Method on GPU Hardware. J. Chem. Theory Comput. 2009, 5, 2371–2377. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernandez, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC: New York, NY, USA, 2023.
- Cockman, M.E.; Lancaster, D.E.; Stolze, I.P.; Hewitson, K.S.; McDonough, M.A.; Coleman, M.L.; Coles, C.H.; Yu, X.; Hay, R.T.; Ley, S.C.; et al. Posttranslational hydroxylation of ankyrin repeats in IκB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc. Natl. Acad. Sci. USA 2006, 103, 14767–14772. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Baldridge, D.; Heeley, J.; Vineyard, M.; Manwaring, L.; Toler, T.L.; Fassi, E.; Fiala, E.; Brown, S.; Goss, C.W.; Willing, M.; et al. The Exome Clinic and the role of medical genetics expertise in the interpretation of exome sequencing results. Genet. Med. 2017, 19, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Courtois, G.; Gilmore, T.D. Mutations in the NF-κB signaling pathway: Implications for human disease. Oncogene 2006, 25, 6831–6843. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Ullah, A.; Shah, A.A.; Syed, F.; Mahmood, A.; Ur Rehman, H.; Khurshid, B.; Samad, A.; Ahmad, W.; Basit, S. Molecular Dynamic Simulation Analysis of a Novel Missense Variant in CYB5R3 Gene in Patients with Methemoglobinemia. Medicina 2023, 59, 379. [Google Scholar] [CrossRef]
- Kaiwar, C.; Zimmermann, M.T.; Ferber, M.J.; Niu, Z.; Urrutia, R.A.; Klee, E.W.; Babovic-Vuksanovic, D. Novel NR2F1 variants likely disrupt DNA binding: Molecular modeling in two cases, review of published cases, genotype-phenotype correlation, and phenotypic expansion of the Bosch-Boonstra-Schaaf optic atrophy syndrome. Cold Spring Harb. Mol. Case Stud. 2017, 3, a002162. [Google Scholar] [CrossRef]
- Muhammad, N.; Hussain, S.I.; Rehman, Z.U.; Khan, S.A.; Jan, S.; Khan, N.; Muzammal, M.; Abbasi, S.W.; Kakar, N.; Rehman, Z.U.; et al. Autosomal recessive variants c.953A>C and c.97-1G>C in NSUN2 causing intellectual disability: A molecular dynamics simulation study of loss-of-function mechanisms. Front. Neurol. 2023, 14, 1168307. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.I.; Stodola, T.J.; De Assuncao, T.M.; Leverence, E.N.; Tripathi, S.; Dsouza, N.R.; Mathison, A.J.; Basel, D.G.; Volkman, B.F.; Smith, B.C.; et al. Molecular mechanics and dynamic simulations of well-known Kabuki syndrome-associated KDM6A variants reveal putative mechanisms of dysfunction. Orphanet J. Rare Dis. 2021, 16, 66. [Google Scholar] [CrossRef]
- Tam, B.; Sinha, S.; Wang, S.M. Combining Ramachandran plot and molecular dynamics simulation for structural-based variant classification: Using TP53 variants as model. Comput. Struct. Biotechnol. J. 2020, 18, 4033–4039. [Google Scholar] [CrossRef]
- Sinha, S.; Wang, S.M. Classification of VUS and unclassified variants in BRCA1 BRCT repeats by molecular dynamics simulation. Comput. Struct. Biotechnol. J. 2020, 18, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.R.C.; Da Silva, A.N.R.; Do Nascimento, S.S.; De Mesquita, J.F. In silico analysis and molecular dynamics simulation of human superoxide dismutase 3 (SOD3) genetic variants. J. Cell Biochem. 2019, 120, 3583–3598. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.; Tyagi, C.; Grover, A.; Goswami, S.K. Molecular modeling and molecular dynamics simulations based structural analysis of the SG2NA protein variants. BMC Res. Notes 2014, 7, 446. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Cai, J.; Li, R.; Wen, C.; Tan, H.; on behalf of the Alzheimer’s Disease Neuroimaging Initiative (ADNI) Database. Rare Variant Analysis and Molecular Dynamics Simulation in Alzheimer’s Disease Identifies Exonic Variants in FLG. Genes 2022, 13, 838. [Google Scholar] [CrossRef] [PubMed]
- Pitsillou, E.; Liang, J.J.; Beh, R.C.; Hung, A.; Karagiannis, T.C. Molecular dynamics simulations highlight the altered binding landscape at the spike-ACE2 interface between the Delta and Omicron variants compared to the SARS-CoV-2 original strain. Comput. Biol. Med. 2022, 149, 106035. [Google Scholar] [CrossRef]
- Mandal, N.; Padhi, A.K.; Rath, S.L. Molecular insights into the differential dynamics of SARS-CoV-2 variants of concern. J. Mol. Graph. Model. 2022, 114, 108194. [Google Scholar] [CrossRef]
- Oliver, G.R.; Zimmermann, M.T.; Klee, E.W.; Urrutia, R.A. “The molecule’s the thing:” the promise of molecular modeling and dynamic simulations in aiding the prioritization and interpretation of genomic testing results. F1000Research 2016, 5, 766. [Google Scholar] [CrossRef]
- Gupta, Y.; Savytskyi, O.V.; Coban, M.; Venugopal, A.; Pleqi, V.; Weber, C.A.; Chitale, R.; Durvasula, R.; Hopkins, C.; Kempaiah, P.; et al. Protein structure-based in-silico approaches to drug discovery: Guide to COVID-19 therapeutics. Mol. Asp. Med. 2023, 91, 101151. [Google Scholar] [CrossRef]
- Selvam, P.; Jain, A.; Abbott, J.; Ahuja, A.S.; Cheema, A.; Bruno, K.A.; Atwal, H.; Forghani, I.; Caulfield, T.; Atwal, P.S. Molecular Modeling and Phenotypic Description of a Patient with a Novel Exonic Deletion of GALNS with Resultant Morquio Syndrome with Two Successful Pregnancies. Mol. Syndromol. 2022, 13, 282–289. [Google Scholar] [CrossRef]
- Richter, J.E., Jr.; Hines, S.; Selvam, P.; Atwal, H.; Farres, H.; Caulfield, T.R.; Atwal, P.S. Clinical description & molecular modeling of novel MAX pathogenic variant causing pheochromocytoma in family, supports paternal parent-of-origin effect. Cancer Genet. 2021, 252–253, 107–110. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD((R))): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef]
- Zhou, X.; Edmonson, M.N.; Wilkinson, M.R.; Patel, A.; Wu, G.; Liu, Y.; Li, Y.; Zhang, Z.; Rusch, M.C.; Parker, M.; et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat. Genet. 2016, 48, 4–6. [Google Scholar] [CrossRef]
- Koch, L. Exploring human genomic diversity with gnomAD. Nat. Rev. Genet. 2020, 21, 448. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaudhri, E.N.; Abbott, J.M.; Islam, N.N.; Weber, C.A.; Coban, M.A.; Bilgili, A.; Squire, J.D.; Mantia, S.; Wierenga, K.J.; Caulfield, T.R. Statistical Mechanics Metrics in Pairing and Parsing In Silico and Phenotypic Data of a Novel Genetic NFκB1 (c.T638A) Variant. Genes 2023, 14, 1855. https://doi.org/10.3390/genes14101855
Chaudhri EN, Abbott JM, Islam NN, Weber CA, Coban MA, Bilgili A, Squire JD, Mantia S, Wierenga KJ, Caulfield TR. Statistical Mechanics Metrics in Pairing and Parsing In Silico and Phenotypic Data of a Novel Genetic NFκB1 (c.T638A) Variant. Genes. 2023; 14(10):1855. https://doi.org/10.3390/genes14101855
Chicago/Turabian StyleChaudhri, Eman N., Jessica M. Abbott, Naeyma N. Islam, Caleb A. Weber, Mathew A. Coban, Ahmet Bilgili, Jacqueline D. Squire, Sarah Mantia, Klaas J. Wierenga, and Thomas R. Caulfield. 2023. "Statistical Mechanics Metrics in Pairing and Parsing In Silico and Phenotypic Data of a Novel Genetic NFκB1 (c.T638A) Variant" Genes 14, no. 10: 1855. https://doi.org/10.3390/genes14101855
APA StyleChaudhri, E. N., Abbott, J. M., Islam, N. N., Weber, C. A., Coban, M. A., Bilgili, A., Squire, J. D., Mantia, S., Wierenga, K. J., & Caulfield, T. R. (2023). Statistical Mechanics Metrics in Pairing and Parsing In Silico and Phenotypic Data of a Novel Genetic NFκB1 (c.T638A) Variant. Genes, 14(10), 1855. https://doi.org/10.3390/genes14101855