
Whole-Exome Sequencing of Pakistani Consanguineous Families Identified Pathogenic Variants in Genes of Intellectual Disability
 ,
,  , ,
, ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
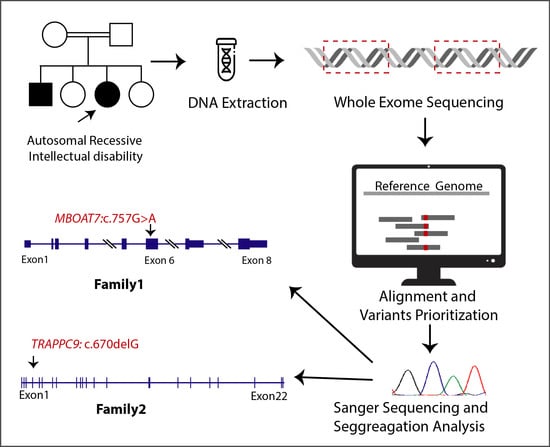
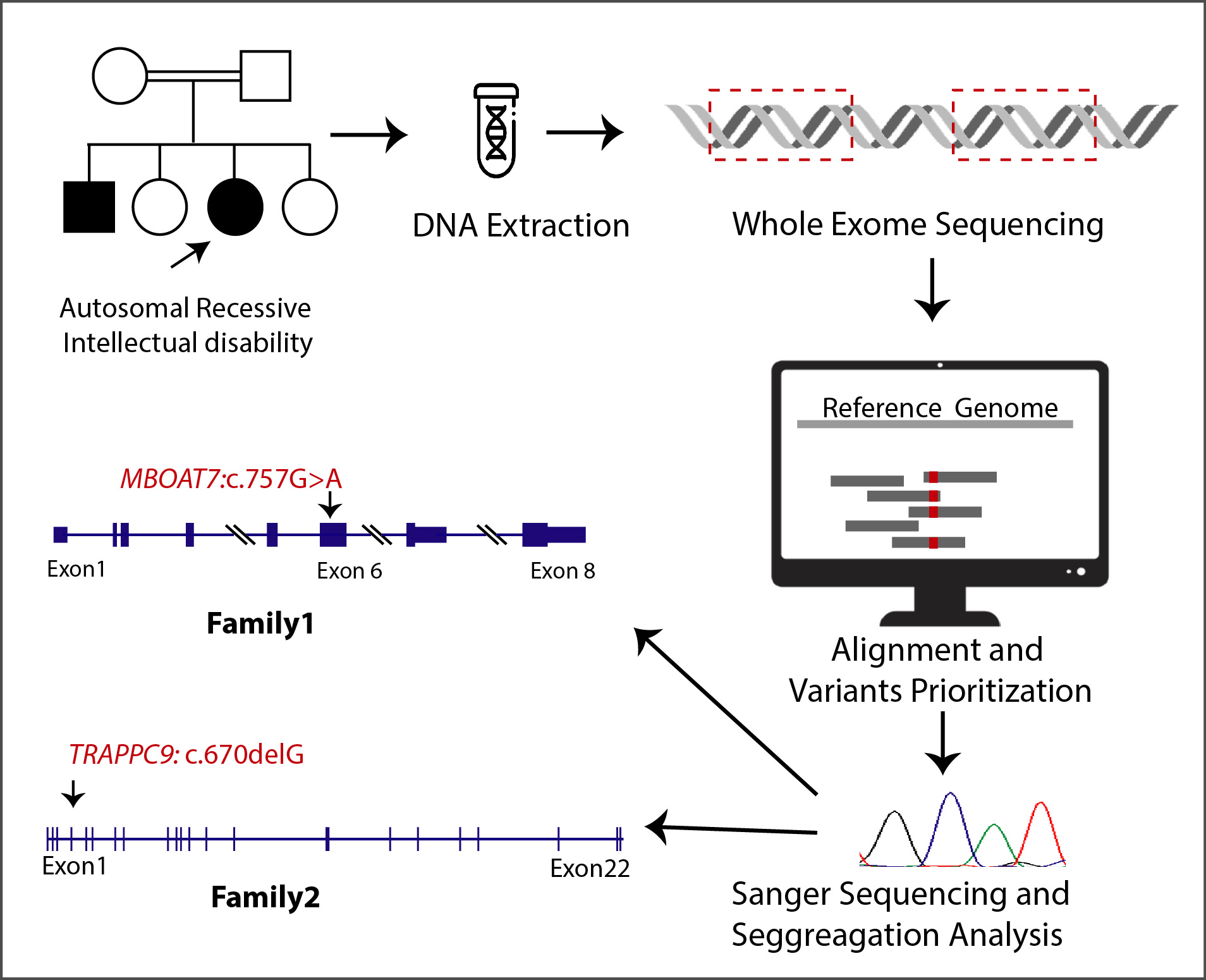
2.2. Whole-Exome Sequencing
2.3. Co-Segregation Analysis
2.4. In Silico Analysis
3. Results
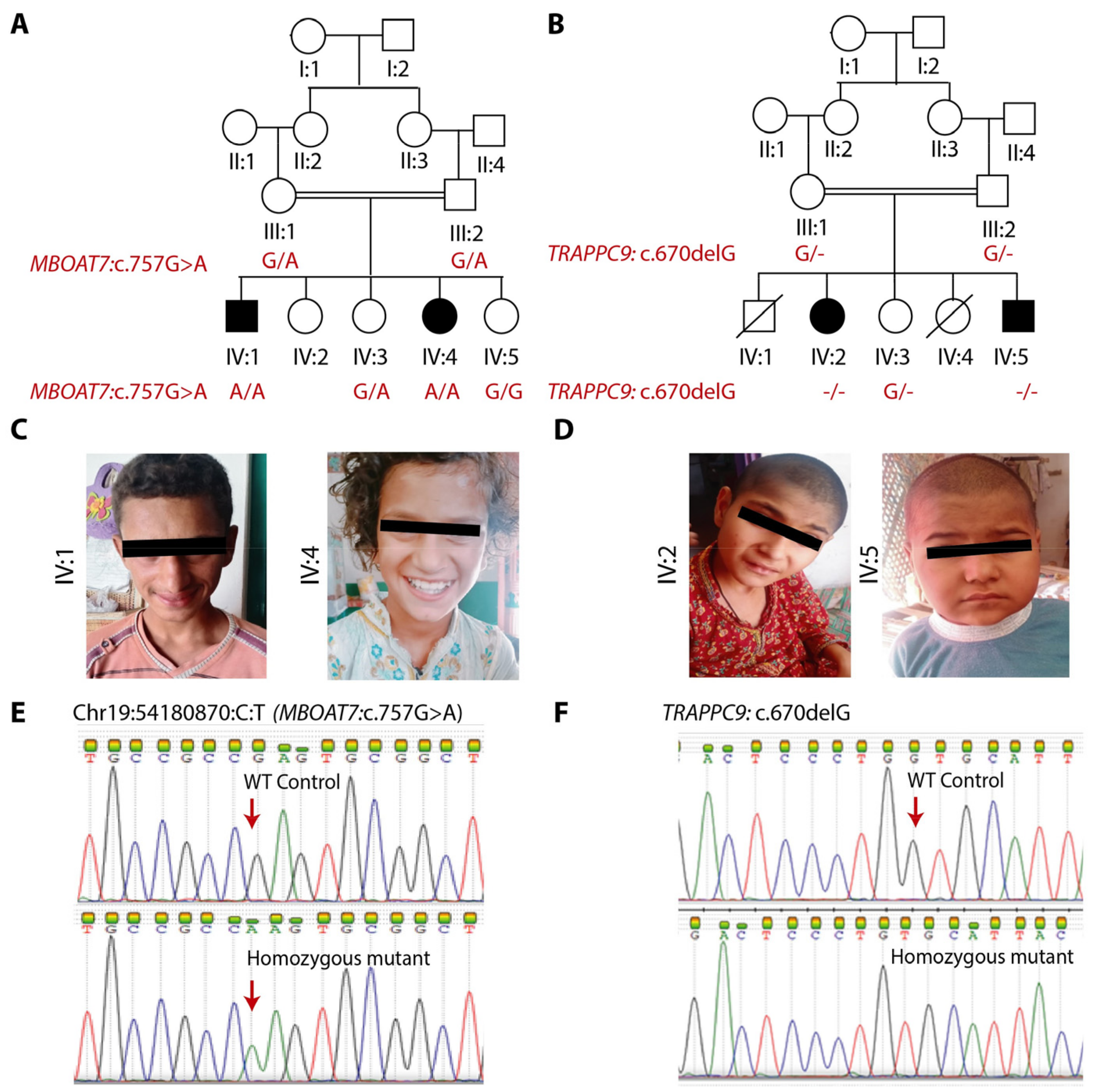
3.1. Clinical Findings
3.2. Genetic Findings
3.3. In Silico Findings
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schalock, R.L.; Borthwick-Duffy, S.A.; Bradley, V.J.; Buntinx, W.H.; Coulter, D.L.; Craig, E.M.; Gomez, S.C.; Lachapelle, Y.; Luckasson, R.; Reeve, A. Intellectual Disability: Definition, Classification, and Systems of Supports, 11th ed.; AAIDD: Washington, DC, USA, 2010. [Google Scholar]
- Schalock, R.L.; Luckasson, R. A systematic approach to subgroup classification in intellectual disability. Intellect. Dev. Disabil. 2015, 53, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Leonard, H.; Wen, X. The epidemiology of mental retardation: Challenges and opportunities in the new millennium. Ment. Retard. Dev. Disabil. Res. Rev. 2002, 8, 117–134. [Google Scholar] [CrossRef]
- Genereaux, D.; van Karnebeek, C.D.; Birch, P.H. Costs of caring for children with an intellectual developmental disorder. Disabil. Health J. 2015, 8, 646–651. [Google Scholar] [CrossRef]
- Shaw-Smith, C.; Redon, R.; Rickman, L.; Rio, M.; Willatt, L.; Fiegler, H.; Firth, H.; Sanlaville, D.; Winter, R.; Colleaux, L. Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J. Med. Genet. 2004, 41, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Vissers, L.E.; de Ligt, J.; Gilissen, C.; Janssen, I.; Steehouwer, M.; de Vries, P.; van Lier, B.; Arts, P.; Wieskamp, N.; del Rosario, M. A de novo paradigm for mental retardation. Nat. Genet. 2010, 42, 1109–1112. [Google Scholar] [CrossRef]
- Hu, H.; Kahrizi, K.; Musante, L.; Fattahi, Z.; Herwig, R.; Hosseini, M.; Oppitz, C.; Abedini, S.S.; Suckow, V.; Larti, F. Genetics of intellectual disability in consanguineous families. Mol. Psychiatry 2019, 24, 1027–1039. [Google Scholar] [CrossRef]
- Vissers, L.E.; Gilissen, C.; Veltman, J.A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 2016, 17, 9. [Google Scholar] [CrossRef]
- Leite, A.J.d.C.; Pinto, I.P.; Leijsten, N.; Ruiterkamp-Versteeg, M.; Pfundt, R.; de Leeuw, N.; da Cruz, A.D.; Minasi, L.B. Diagnostic yield of patients with undiagnosed intellectual disability, global developmental delay and multiples congenital anomalies using karyotype, microarray analysis, whole exome sequencing from Central Brazil. PLoS ONE 2022, 17, e0266493. [Google Scholar] [CrossRef]
- Han, J.Y.; Lee, I.G. Genetic tests by next-generation sequencing in children with developmental delay and/or intellectual disability. Clin. Exp. Pediatr. 2020, 63, 195. [Google Scholar] [CrossRef]
- Hussain, M.S.; Baig, S.M.; Neumann, S.; Nürnberg, G.; Farooq, M.; Ahmad, I.; Alef, T.; Hennies, H.C.; Technau, M.; Altmüller, J. A truncating mutation of CEP135 causes primary microcephaly and disturbed centrosomal function. Am. J. Hum. Genet. 2012, 90, 871–878. [Google Scholar] [CrossRef]
- Caddeo, A.; Jamialahmadi, O.; Solinas, G.; Pujia, A.; Mancina, R.M.; Pingitore, P.; Romeo, S. MBOAT7 is anchored to endomembranes by six transmembrane domains. J. Struct. Biol. 2019, 206, 349–360. [Google Scholar] [CrossRef]
- Pérez-Chacón, G.; Astudillo, A.M.; Balgoma, D.; Balboa, M.A.; Balsinde, J. Control of free arachidonic acid levels by phospholipases A2 and lysophospholipid acyltransferases. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2009, 1791, 1103–1113. [Google Scholar] [CrossRef]
- Johansen, A.; Rosti, R.O.; Musaev, D.; Sticca, E.; Harripaul, R.; Zaki, M.; Çağlayan, A.O.; Azam, M.; Sultan, T.; Froukh, T. Mutations in MBOAT7, encoding lysophosphatidylinositol acyltransferase I, lead to intellectual disability accompanied by epilepsy and autistic features. Am. J. Hum. Genet. 2016, 99, 912–916. [Google Scholar] [CrossRef]
- Lee, J.; Shamim, A.; Park, J.; Jang, J.; Kim, J.H.; Kwon, J.; Kim, J.-W.; Kim, K.K.; Lee, J. Functional and structural changes in the membrane-bound Oacyltransferase family member 7 (MBOAT7) protein. Front. Neurol. 2022, 13, 836954. [Google Scholar] [CrossRef]
- Ma, D.; Wang, Z.; Merrikh, C.N.; Lang, K.S.; Lu, P.; Li, X.; Merrikh, H.; Rao, Z.; Xu, W. Crystal structure of a membrane-bound O-acyltransferase. Nature 2018, 562, 286–290. [Google Scholar] [CrossRef]
- Barrowman, J.; Bhandari, D.; Reinisch, K.; Ferro-Novick, S. TRAPP complexes in membrane traffic: Convergence through a common Rab. Nat. Rev. Mol. Cell Biol. 2010, 11, 759–763. [Google Scholar] [CrossRef]
- Radenkovic, S.; Martinelli, D.; Zhang, Y.; Preston, G.J.; Maiorana, A.; Terracciano, A.; Dentici, M.L.; Pisaneschi, E.; Novelli, A.; Ranatunga, W. TRAPPC9-CDG: A novel congenital disorder of glycosylation with dysmorphic features and intellectual disability. Genet. Med. 2022, 24, 894–904. [Google Scholar] [CrossRef]
- Bolat, H.; Ünsel-Bolat, G.; Derin, H.; Şen, A.; Ceylaner, S. Distinct Autism Spectrum Disorder Phenotype and Hand-Flapping Stereotypes: Two Siblings with Novel Homozygous Mutation in TRAPPC9 Gene and Literature Review. Mol. Syndromol. 2022, 13, 263–269. [Google Scholar] [CrossRef]
- Alvarez-Mora, M.I.; Corominas, J.; Gilissen, C.; Sanchez, A.; Madrigal, I.; Rodriguez-Revenga, L. Novel Compound Heterozygous Mutation in TRAPPC9 Gene: The relevance of whole genome sequencing. Genes 2021, 12, 557. [Google Scholar] [CrossRef]
- Ke, Y.; Weng, M.; Chhetri, G.; Usman, M.; Li, Y.; Yu, Q.; Ding, Y.; Wang, Z.; Wang, X.; Sultana, P. Trappc9 deficiency in mice impairs learning and memory by causing imbalance of dopamine D1 and D2 neurons. Sci. Adv. 2020, 6, eabb7781. [Google Scholar] [CrossRef] [PubMed]
- Usman, M.; Li, Y.; Ke, Y.; Chhetri, G.; Islam, M.A.; Wang, Z.; Li, X. Trappc9 Deficiency Impairs the Plasticity of Stem Cells. Int. J. Mol. Sci. 2022, 23, 4900. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Family IDs | Family A | Family B | ||
|---|---|---|---|---|
| Mutation | MBOAT7 (NM_024298.4: c.757G>A; p.(Glu253Lys)) | TRAPPC9 (NM_001160372.3: c.670delG; p.(Val224Cysfs*13) | ||
| Patient ID | IV:1 | IV:4 | IV:2 | IV:5 |
| Sex | Male | Female | Female | Male |
| Age at examination | 16 years | 10 years | 10 years | 4 years |
| Height | 175.25 cm | 137.20 cm (−0.3 SD) | 121.90 cm (−3 SD) | 86.35 cm (−4 SD) |
| HC | 50.10 cm (−4 SD) | 53.35 cm (+1 SD) | 45.72 cm (−5 SD) | 43.20 cm (−7 SD) |
| Intellectual disability | Severe | Severe | Severe | Severe |
| Self-care | No | No | No | No |
| Self-feed | No | No | No | No |
| Gestational history | Unremarkable | Unremarkable | Unremarkable | Unremarkable |
| Sitting | 1 year | 2 years | 2 years | 2 years |
| Walking | 3 years | 3.5 years | 5 years | Not attained |
| Speech Impairment | Yes | Yes | Yes | Yes |
| Visual Impairment | No | No | No | No |
| Ambulation Difficulty | No | No | No | No |
| Facial Dysmorphism | No | No | No | No |
| Aggressive | Yes | Yes | N/A | N/A |
| Seizures | No | Yes, started at the age of 5 months | Yes, onset at the age of 4 years | Yes, onset at the age of 1.5 years |
| Skin anomalies | No | No | No | No |
| Prediction Tool | Prediction and Score |
|---|---|
| gnomAD frequency | 1/152, 184 (0.000006571) |
| CADD | 27.1 |
| PolyPhen-2 | Probably damaging (0.999) |
| DANN | Pathogenic (0.9993) |
| EIGEN | Pathogenic (0.7627) |
| SIFT | Deleterious |
| Mutation Taster | Disease-causing (56) |
| Polyphen-2 | Probably damaging (1.000) |
| Conservation Scores | phyloP100: 4.065 |
| EIGEN PC | Pathogenic (0.7029) |
| PROVEAN | Uncertain (3.28, 3.46, 3.05, 3.03) |
| ACMG Classification | Uncertain significance (PM1, PP3 and BP1) |
| SNAP2 | Neutral −41 (72%) |
| DEOGEN2 | Uncertain (Damaging, Tolerated) 0.5828, 0.4976 |
| MetaRNN | Pathogenic |
| FATHMM-MKL | Pathogenic (0.9838) |
| FATHMM-XF | Uncertain (0.8913) |
| MutPred | Pathogenic (0.84) |
| PANTHER | (Probably damaging) (0.74) |
| REVEL | Pathogenic (0.7459) |
| BayesDel addAF | Pathogenic (addAF score 0.2137) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asif, M.; Anayat, M.; Tariq, F.; Noureen, T.; Din, G.N.U.; Becker, C.; Becker, K.; Thiele, H.; Makhdoom, E.u.H.; Shaiq, P.A.; et al. Whole-Exome Sequencing of Pakistani Consanguineous Families Identified Pathogenic Variants in Genes of Intellectual Disability. Genes 2023, 14, 48. https://doi.org/10.3390/genes14010048
Asif M, Anayat M, Tariq F, Noureen T, Din GNU, Becker C, Becker K, Thiele H, Makhdoom EuH, Shaiq PA, et al. Whole-Exome Sequencing of Pakistani Consanguineous Families Identified Pathogenic Variants in Genes of Intellectual Disability. Genes. 2023; 14(1):48. https://doi.org/10.3390/genes14010048
Chicago/Turabian StyleAsif, Maria, Maryam Anayat, Faiza Tariq, Tanzeela Noureen, Ghulam Naseer Ud Din, Christian Becker, Kerstin Becker, Holger Thiele, Ehtisham ul Haq Makhdoom, Pakeeza Arzoo Shaiq, and et al. 2023. "Whole-Exome Sequencing of Pakistani Consanguineous Families Identified Pathogenic Variants in Genes of Intellectual Disability" Genes 14, no. 1: 48. https://doi.org/10.3390/genes14010048
APA StyleAsif, M., Anayat, M., Tariq, F., Noureen, T., Din, G. N. U., Becker, C., Becker, K., Thiele, H., Makhdoom, E. u. H., Shaiq, P. A., Baig, S. M., Nürnberg, P., Hussain, M. S., Raja, G. K., & Abdullah, U. (2023). Whole-Exome Sequencing of Pakistani Consanguineous Families Identified Pathogenic Variants in Genes of Intellectual Disability. Genes, 14(1), 48. https://doi.org/10.3390/genes14010048