
Effective smMIPs-Based Sequencing of Maculopathy-Associated Genes in Stargardt Disease Cases and Allied Maculopathies from the UK

 , , , , , , , and
, , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. smMIPs Design
2.2. Patient Cohort
2.3. Sample Preparation
2.4. Variant Calling and Annotation
2.5. Variant Prioritisation
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perez-Carro, R.; Corton, M.; Sánchez-Navarro, I.; Zurita, O.; Sanchez-Bolivar, N.; Sánchez-Alcudia, R.; Lelieveld, S.H.; Aller, E.; Lopez-Martinez, M.A.; López-Molina, M.I.; et al. Panel-Based NGS Reveals Novel Pathogenic Mutations in Autosomal Recessive Retinitis Pigmentosa. Sci. Rep. 2016, 6, 19531. [Google Scholar] [CrossRef] [PubMed]
- Neveling, K.; Collin, R.W.J.; Gilissen, C.; Van Huet, R.A.C.; Visser, L.; Kwint, M.P.; Gijsen, S.J.; Zonneveld, M.N.; Wieskamp, N.; De Ligt, J.; et al. Next-Generation Genetic Testing for Retinitis Pigmentosa. Hum. Mutat. 2012, 33, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Riera, M.; Navarro, R.; Ruiz-Nogales, S.; Méndez, P.; Burés-Jelstrup, A.; Corcóstegui, B.; Pomares, E. Whole Exome Sequencing Using Ion Proton System Enables Reliable Genetic Diagnosis of Inherited Retinal Dystrophies. Sci. Rep. 2017, 7, 42078. [Google Scholar] [CrossRef] [PubMed]
- Dockery, A.; Whelan, L.; Humphries, P.; Jane Farrar, G. Next-Generation Sequencing Applications for Inherited Retinal Diseases. Int. J. Mol. Sci. 2021, 22, 5684. [Google Scholar] [CrossRef]
- Ge, Z.; Bowles, K.; Goetz, K.; Scholl, H.P.N.; Wang, F.; Wang, X.; Xu, S.; Wang, K.; Wang, H.; Chen, R. NGS-Based Molecular Diagnosis of 105 EyeGENE ® Probands with Retinitis Pigmentosa. Sci. Rep. 2015, 5, 18287. [Google Scholar] [CrossRef]
- O’Sullivan, J.; Mullaney, B.G.; Bhaskar, S.S.; Dickerson, J.E.; Hall, G.; O’Grady, A.; Webster, A.; Ramsden, S.C.; Black, G.C. A Paradigm Shift in the Delivery of Services for Diagnosis of Inherited Retinal Disease. J. Med. Genet. 2012, 49, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, F.; Wang, H.; Li, Y.; Alexander, S.; Wang, K.; Willoughby, C.E.; Zaneveld, J.E.; Jiang, L.; Soens, Z.T.; et al. Next-Generation Sequencing-Based Molecular Diagnosis of 82 Retinitis Pigmentosa Probands from Northern Ireland. Hum. Genet. 2015, 134, 217–230. [Google Scholar] [CrossRef]
- Consugar, M.B.; Navarro-Gomez, D.; Place, E.M.; Bujakowska, K.M.; Sousa, M.E.; Fonseca-Kelly, Z.D.; Taub, D.G.; Janessian, M.; Wang, D.Y.; Au, E.D.; et al. Panel-Based Genetic Diagnostic Testing for Inherited Eye Diseases Is Highly Accurate and Reproducible, and More Sensitive for Variant Detection, than Exome Sequencing. Genet. Med. 2015, 17, 253–261. [Google Scholar] [CrossRef]
- Audo, I.; Bujakowska, K.M.; Léveillard, T.; Mohand-Sad, S.; Lancelot, M.E.; Germain, A.; Antonio, A.; Michiels, C.; Saraiva, J.P.; Letexier, M.; et al. Development and Application of a Next-Generation-Sequencing (NGS) Approach to Detect Known and Novel Gene Defects Underlying Retinal Diseases. Orphanet J. Rare Dis. 2012, 7, 8. [Google Scholar] [CrossRef]
- Haer-Wigman, L.; Van Zelst-Stams, W.A.G.; Pfundt, R.; Van Den Born, L.I.; Klaver, C.C.W.; Verheij, J.B.G.M.; Hoyng, C.B.; Breuning, M.H.; Boon, C.J.F.; Kievit, A.J.; et al. Diagnostic Exome Sequencing in 266 Dutch Patients with Visual Impairment. Eur. J. Hum. Genet. 2017, 25, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Ellingford, J.M.; Barton, S.; Bhaskar, S.; O’Sullivan, J.; Williams, S.G.; Lamb, J.A.; Panda, B.; Sergouniotis, P.I.; Gillespie, R.L.; Daiger, S.P.; et al. Molecular Findings from 537 Individuals with Inherited Retinal Disease. J. Med. Genet. 2016, 53, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Bahr, A.; Bähr, L.; Fleischhauer, J.; Zinkernagel, M.S.; Winkler, N.; Barthelmes, D.; Berger, L.; Gerth-Kahlert, C.; Neidhardt, J.; et al. Next Generation Sequencing Based Identification of Disease-Associated Mutations in Swiss Patients with Retinal Dystrophies. Sci. Rep. 2016, 6, 28755. [Google Scholar] [CrossRef]
- Ma, D.J.; Lee, H.S.; Kim, K.; Choi, S.; Jang, I.; Cho, S.H.; Yoon, C.K.; Lee, E.K.; Yu, H.G. Whole-Exome Sequencing in 168 Korean Patients with Inherited Retinal Degeneration. BMC Med. Genom. 2021, 14, 74. [Google Scholar] [CrossRef]
- Carss, K.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Ellingford, J.M.; Barton, S.; Bhaskar, S.; Williams, S.G.; Sergouniotis, P.I.; O’Sullivan, J.; Lamb, J.A.; Perveen, R.; Hall, G.; Newman, W.G.; et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology 2016, 123, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.L.; Pierce, E.A.; Laster, A.M.; Daiger, S.P.; Birch, D.G.; Ash, J.D.; Iannaccone, A.; Flannery, J.G.; Sahel, J.A.; Zack, D.J.; et al. Inherited Retinal Degenerations: Current Landscape and Knowledge Gaps. Transl. Vis. Sci. Technol. 2018, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.A.J.; Zhu, J.; Wynne, N.; Dockery, A.; Cairns, R.M.; Duignan, E.; Whelan, L.; Malone, C.P.; Dempsey, H.; Collins, K.; et al. Target 5000: A Standardized All-Ireland Pathway for the Diagnosis and Management of Inherited Retinal Degenerations. Orphanet J. Rare Dis. 2021, 16, 200. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Cornelis, S.S.; Del Pozo-Valero, M.; Whelan, L.; Runhart, E.H.; Mishra, K.; Bults, F.; AlSwaiti, Y.; AlTalbishi, A.; De Baere, E.; et al. Resolving the Dark Matter of ABCA4 for 1054 Stargardt Disease Probands through Integrated Genomics and Transcriptomics. Genet. Med. 2020, 22, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Hardenbol, P.; Banér, J.; Jain, M.; Nilsson, M.; Namsaraev, E.A.; Karlin-Neumann, G.A.; Fakhrai-Rad, H.; Ronaghi, M.; Willis, T.D.; Landegren, U.; et al. Multiplexed Genotyping with Sequence-Tagged Molecular Inversion Probes. Nat. Biotechnol. 2003, 21, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Schueler, M.; Halbritter, J.; Phelps, I.G.; Braun, D.A.; Otto, E.A.; Porath, J.D.; Gee, H.Y.; Shendure, J.; O’Roak, B.J.; Lawson, J.A.; et al. Large-Scale Targeted Sequencing Comparison Highlights Extreme Genetic Heterogeneity in Nephronophthisis-Related Ciliopathies. J. Med. Genet. 2016, 53, 208–214. [Google Scholar] [CrossRef]
- Weisschuh, N.; Feldhaus, B.; Khan, M.I.; Cremers, F.P.M.; Kohl, S.; Wissinger, B.; Zobor, D. Molecular and Clinical Analysis of 27 German Patients with Leber Congenital Amaurosis. PLoS ONE 2018, 13, e0205380. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Hitti-Malin, R.J.; Dhaenens, C.; Panneman, D.M.; Corradi, Z.; Khan, M.; den Hollander, A.I.; Farrar, G.J.; Gilissen, C.; Hoischen, A.; van de Vorst, M.; et al. Using Single Molecule Molecular Inversion Probes as a Cost-Effective, High-Throughput Sequencing Approach to Target All Genes and Loci Associated with Macular Diseases. Hum. Mutat. 2022, 43, 2234–2250. [Google Scholar] [CrossRef] [PubMed]
- Turner, E.H.; Lee, C.; Ng, S.B.; Nickerson, D.A.; Shendure, J. Massively Parallel Exon Capture and Library-Free Resequencing across 16 Genomes. Nat. Methods 2009, 6, 315–316. [Google Scholar] [CrossRef] [PubMed]
- Hiatt, J.B.; Pritchard, C.C.; Salipante, S.J.; O’Roak, B.J.; Shendure, J. Single Molecule Molecular Inversion Probes for Targeted, High-Accuracy Detection of Low-Frequency Variation. Genome Res. 2013, 23, 843–854. [Google Scholar] [CrossRef]
- Rowe, L.R.; Thaker, H.M.; Opitz, J.M.; Schiffman, J.D.; Haddadin, Z.M.; Erickson, L.K.; South, S.T. Molecular Inversion Probe Array for the Genetic Evaluation of Stillbirth Using Formalin-Fixed, Paraffin-Embedded Tissue. J. Mol. Diagn. 2013, 15, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Shu, L.; Zhong, J.; Pan, H.; Guo, J.; Sun, Q.; Yan, X.; Tang, B.; Xu, Q. Association of HIF1A and Parkinson’s Disease in a Han Chinese Population Demonstrated by Molecular Inversion Probe Analysis. Neurol. Sci. 2019, 40, 1927–1931. [Google Scholar] [CrossRef] [PubMed]
- Jahromi, M.S.; Putnam, A.R.; Druzgal, C.; Wright, J.; Spraker-Perlman, H.; Kinsey, M.; Zhou, H.; Boucher, K.M.; Randall, R.L.; Jones, K.B.; et al. Molecular Inversion Probe Analysis Detects Novel Copy Number Alterations in Ewing Sarcoma. Cancer Genet. 2012, 205, 391–404. [Google Scholar] [CrossRef]
- Schiffman, J.D.; Wang, Y.; McPherson, L.A.; Welch, K.; Zhang, N.; Davis, R.; Lacayo, N.J.; Dahl, G.V.; Faham, M.; Ford, J.M.; et al. Molecular Inversion Probes Reveal Patterns of 9p21 Deletion and Copy Number Aberrations in Childhood Leukemia. Cancer Genet. Cytogenet. 2009, 193, 9. [Google Scholar] [CrossRef]
- Xu, H.L.; Xu, W.H.; Cai, Q.; Feng, M.; Long, J.; Zheng, W.; Xiang, Y.B.; Shu, X.O. Polymorphisms and Haplotypes in the Caspase 3, 7, and 8 Genes and Risk of Endometrial Cancer: A Population-Based, Case-Control Study in a Chinese Population. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2114. [Google Scholar] [CrossRef]
- Suzuki, O.; Dong, O.M.; Howard, R.M.; Wiltshire, T. Characterizing the Pharmacogenome Using Molecular Inversion Probes for Targeted Next-Generation Sequencing. Pharmacogenomics 2019, 20, 1005–1020. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Cornelis, S.S.; Khan, M.I.; Elmelik, D.; Manders, E.; Bakker, S.; Derks, R.; Neveling, K.; van de Vorst, M.; Gilissen, C.; et al. Cost-effective Molecular Inversion Probe-based ABCA4 Sequencing Reveals Deep-intronic Variants in Stargardt Disease. Hum. Mutat. 2019, 40, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- Runhart, E.H.; Sangermano, R.; Cornelis, S.S.; Verheij, J.B.G.M.; Plomp, A.S.; Boon, C.J.F.; Lugtenberg, D.; Roosing, S.; Bax, N.M.; Blokland, E.A.W.; et al. The Common ABCA4 Variant p.Asn1868ile Shows Nonpenetrance and Variable Expression of Stargardt Disease When Present in Trans with Severe Variants. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3220–3231. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, S.S.; Runhart, E.H.; Bauwens, M.; Corradi, Z.; De Baere, E.; Roosing, S.; Haer-Wigman, L.; Dhaenens, C.-M.; Vulto-Van Silfhout, A.T.; Cremers, F.P.M. Personalized Genetic Counseling for Stargardt Disease: Offspring Risk Estimates Based on Variant Severity. Am. J. Hum. Genet. 2022, 109(3), 498–507. [Google Scholar] [CrossRef] [PubMed]
- Cremers, F.P.M.; Lee, W.; Collin, R.W.J.; Allikmets, R. Clinical Spectrum, Genetic Complexity and Therapeutic Approaches forretinal Disease Caused by ABCA4 Mutations. Prog. Retin. Eye Res. 2020, 79, 100861. [Google Scholar] [CrossRef] [PubMed]
- Sheck, L.H.N.; Esposti, S.D.; Mahroo, O.A.; Arno, G.; Pontikos, N.; Wright, G.; Webster, A.R.; Khan, K.N.; Michaelides, M. Panel-Based Genetic Testing for Inherited Retinal Disease Screening 176 Genes. Mol. Genet. Genom. Med. 2021, 9, e1663. [Google Scholar] [CrossRef] [PubMed]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; den Dunnen, J.T. LOVD v.2.0: The next Generation in Gene Variant Databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- Cornelis, S.S.; Bax, N.M.; Zernant, J.; Allikmets, R.; Fritsche, L.G.; den Dunnen, J.T.; Ajmal, M.; Hoyng, C.B.; Cremers, F.P.M. In Silico Functional Meta-Analysis of 5962 ABCA4 Variants in 3928 Retinal Dystrophy Cases. Hum. Mutat. 2017, 38, 400–408. [Google Scholar] [CrossRef]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of Nonneutral Substitution Rates on Mammalian Phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’roak, B.J.; Cooper, G.M.; Shendure, J. A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Grantham, R. Amino Acid Difference Formula to Help Explain Protein Evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Panneman, D.M.; Hitti-Malin, R.J.; Holtes, L.K.; de Bruijn, S.E.; Reurink, J.; Boonen, E.G.M.; Khan, M.I.; Ali, M.; Andréasson, S.; De Baere, E.; et al. Cost-Effective Sequence Analysis of 113 Genes in 1,192 Probands with Retinitis Pigmentosa and Leber Congenital Amaurosis. medRxiv 2022, 2022.11.24.22282656v2. [Google Scholar] [CrossRef]
- Zernant, J.; Lee, W.; Collison, F.T.; Fishman, G.A.; Sergeev, Y.V.; Schuerch, K.; Sparrow, J.R.; Tsang, S.H.; Allikmets, R. Frequent Hypomorphic Alleles Account for a Significant Fraction of ABCA4 Disease and Distinguish It from Age-Related Macular Degeneration. J. Med. Genet. 2017, 54, 404. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, Y.; Jiang, L.; Karan, G.; Moshfeghi, D.M.; O’Connor, S.T.; Li, X.; Yu, Z.; Lewis, H.; Zack, D.J.; et al. A Novel RDS/Peripherin Gene Mutation Associated with Diverse Macular Phenotypes. Ophthalmic Genet. 2004, 25, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ge, X.; Shi, W.; Huang, P.; Min, Q.; Li, M.; Yu, X.; Wu, Y.; Zhao, G.; Tong, Y.; et al. Molecular Diagnosis of Putative Stargardt Disease by Capture next Generation Sequencing. PLoS ONE 2014, 9, e95528. [Google Scholar] [CrossRef] [PubMed]
- Imani, S.; Cheng, J.; Shasaltaneh, M.D.; Wei, C.; Yang, L.; Fu, S.; Zou, H.; Khan, M.A.; Zhang, X.; Chen, H.; et al. Genetic Identification and Molecular Modeling Characterization Reveal a Novel PROM1 Mutation in Stargardt4-like Macular Dystrophy. Oncotarget 2017, 9, 122–141. [Google Scholar] [CrossRef] [PubMed]
- Kniazeva, M.; Chiang, M.F.; Morgan, B.; Anduze, A.L.; Zack, D.J.; Han, M.; Zhang, K. A New Locus for Autosomal Dominant Stargardt-like Disease Maps to Chromosome 4. Am. J. Hum. Genet. 1999, 64, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.M.; El-Asrag, M.; Parry, D.A.; Morgan, J.E.; Logan, C.V.; Carr, I.M.; Sheridan, E.; Charlton, R.; Johnson, C.A.; Taylor, G.; et al. Mutation Screening of Retinal Dystrophy Patients by Targeted Capture from Tagged Pooled DNAs and Next Generation Sequencing. PLoS ONE 2014, 9, e104281. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.M.; Crinnion, L.A.; Hewitt, S.; Bates, J.; Robinson, R.; Carr, I.M.; Sheridan, E.; Adlard, J.; Bonthron, D.T. Cas9-Based Enrichment and Single-Molecule Sequencing for Precise Characterization of Genomic Duplications. Lab. Investig. 2019, 100, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Ruddle, J.B.; Ebenezer, N.D.; Kearns, L.S.; Mulhall, L.E.; Mackey, D.A.; Hardcastle, A.J. RPGR ORF15 Genotype and Clinical Variability of Retinal Degeneration in an Australian Population. Br. J. Ophthalmol. 2009, 93, 1151–1154. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| ID | Gene | Allele 1 | Allele 2 | ||||
|---|---|---|---|---|---|---|---|
| cDNA | Protein | ACMG | cDNA | Protein | ACMG | ||
| 1337 | ABCA4 | c.5603A>T(;)5819T>C | p.(Asn1868Ile)(;)(Leu1940Pro) | VUS,LP | c.6817-2A>C | p.(?) | P |
| 1746 | ABCA4 | c.[2588G>C;5603A>T] | p.[[Gly863Ala,Gly863del];(Asn1868Ile)] | P | c.[5461-10T>C;5603A>T] | p.[[Thr1821Aspfs*6,Thr1821Valfs*13]; (Asn1868Ile)] | P |
| 1808 | ABCA4 | c.[5461-10T>C;5603A>T] | p.[[Thr1821Aspfs*6,Thr1821Valfs*13]; (Asn1868Ile)] | P | c.5882G>A | p.(Gly1961Glu) | P |
| 2469 | ABCA4 | c.[2588G>C;5603A>T] | p.[[Gly863Ala,Gly863del];(Asn1868Ile)] | P | c.[5461-10T>C;5603A>T] | p.[[Thr1821Aspfs*6,Thr1821Valfs*13]; (Asn1868Ile)] | P |
| 2843 | ABCA4 | c.4016G>A(;)5313-1_5313del | p.(Cys1339Tyr)(;)(?) | VUS,P | c.6088C>T | p.(Arg2030*) | P |
| 3536 | ABCA4 | c.3113C>T | p.(Ala1038Val) | P | c.1906C>T | p.(Gln636*) | P |
| 3616 | ABCA4 | c.4469G>A | p.(Cys1490Tyr) | P | c.5603A>T | p.(Asn1868Ile) | VUS |
| PRPH2 | c.623G>A | p.(Gly208Asp) | P | -- | -- | -- | |
| 3656 # | ABCA4 | c.4139C>T | p.(Pro1380Leu) | P | c.5882G>A | p.(Gly1961Glu) | P |
| 4126 | ABCA4 | c.4774-27T>C(;) 5196+1137G>A | p.[=;Gly1592Alafs*113](;) [=;Met1733Glufs*78] | LB, P | c.[5461-10T>C;5603A>T] | p.[[Thr1821Aspfs*6,Thr1821Valfs*13]; (Asn1868Ile)] | P |
| 5219 | ABCA4 | c.634C>T | p.(Arg212Cys) | P | c.[5461-10T>C;5603A>T] | p.[[Thr1821Aspfs*6,Thr1821Valfs*13]; (Asn1868Ile)] | P |
| 5270 | ABCA4 | c.1906C>T | p.(Gln636*) | P | c.[2588G>C;5603A>T] | p.[[Gly863Ala,Gly863del];(Asn1868Ile)] | P |
| 5349 | ABCA4 | c.1906C>T | p.(Gln636*) | P | c.5603A>T | p.(Asn1868Ile) | VUS |
| 5604 | ABCA4 | c.4577C>T(;)4469G>A | p.(Thr1526Met)(;)(Cys1490Tyr) | P,P | c.5603A>T | p.(Asn1868Ile) | VUS |
| 5607 | ABCA4 | c.3259G>A | p.(Glu1087Lys) | P | c.6089G>A | p.(Arg2030Gln) | P |
| 5608 | ABCA4 | c.[2588G>C;5603A>T] | p.[[Gly863Ala,Gly863del];(Asn1868Ile)] | P | c.[5461-10T>C;5603A>T] | p.[[Thr1821Aspfs*6,Thr1821Valfs*13]; (Asn1868Ile)] | P |
| 5609 | ABCA4 | c.4139C>T | p.(Pro1380Leu) | P | c.4139C>T | p.(Pro1380Leu) | P |
| 5851 | ABCA4 | c.1282del | p.(Val428Serfs*7) | P | c.6320G>A | p.(Arg2107His) | P |
| 5852 | ABCA4 | c.[2588G>C;5603A>T] | p.[[Gly863Ala,Gly863del];(Asn1868Ile)] | P | c.[5461-10T>C;5603A>T] | p.[[Thr1821Aspfs*6,Thr1821Valfs*13]; (Asn1868Ile)] | P |
| 5853 | ABCA4 | c.365_366insCA | p.(Gly123Metfs*32) | P | c.5560G>T(;)5603A>T(;)5882G>A | p.(Val1854Leu)(;)(Asn1868Ile)(;) (Gly1961Glu) | LP,VUS,P |
| 5854 | ABCA4 | c.4195G>A | p.(Glu1399Lys) | LP | c.5318C>T | p.(Ala1773Val) | P |
| 5857 # | ABCA4 | c.6229C>T | p.(Arg2077Trp) | P | c.6229C>T | p.(Arg2077Trp) | P |
| 5860 | ABCA4 | c.4326C>A | p.(Asn1442Lys) | LP | c.5882G>A | p.(Gly1961Glu) | P |
| 5861 | ABCA4 | c.5714+5G>A | p.[=,Glu1863Leufs*33] | P | c.[5461-10T>C;5603A>T] | p.[[Thr1821Aspfs*6,Thr1821Valfs*13]; (Asn1868Ile)] | VUS, P |
| 5862 | ABCA4 | c.1906C>T | p.(Gln636*) | P | c.4577C>T | p.(Thr1526Met) | P |
| 5863 | ABCA4 | c.[2588G>C;5603A>T] | p.[[Gly863Ala,Gly863del];(Asn1868Ile)] | P | c.4537del | p.(Gln1513Argfs*13) | P |
| 5864 | ABCA4 | c.1253T>C | p.(Phe418Ser) | P | c.1317G>A | p.(Trp439*) | P |
| 5865 | ABCA4 | c.1906C>T | p.(Gln636*) | P | c.5603A>T | p.(Asn1868Ile) | VUS |
| 3670 | BEST1 | c.728C>T | p.(Ala243Val) | P | -- | -- | -- |
| 3654 | BEST1 | c.889C>T | p.(Pro297Ser) | P | -- | -- | -- |
| 4030 | C1QTNF5 | c.489C>G | p.(Ser163Arg) | P | -- | -- | -- |
| 5258 | CRB1 | c.249T>A | p.(Tyr83*) | LP | c.2506C>A | p.(Pro836Thr) | LP |
| 3615 | PROM1 | c.1117C>T | p.(Arg373Cys) | P | -- | -- | -- |
| 5610 | PROM1 | c.1117C>T | p.(Arg373Cys) | P | -- | -- | -- |
| 3798 | PRPH2 | c.638G>A | p.(Cys213Tyr) | P | -- | -- | -- |
| 4767 # | PRPH2 | c.394del | p.(Gln132Lysfs*7) | P | -- | -- | -- |
| 5855 | PRPH2 | c.291G>A | p.(Trp97*) | LP | -- | -- | -- |
| ID | Clinical data | Genetic data | Match to Model | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Age at Grading | Grade | Diagnosis | Allele 1 | Severity | Allele 2 | Severity | Diagnosis | ||
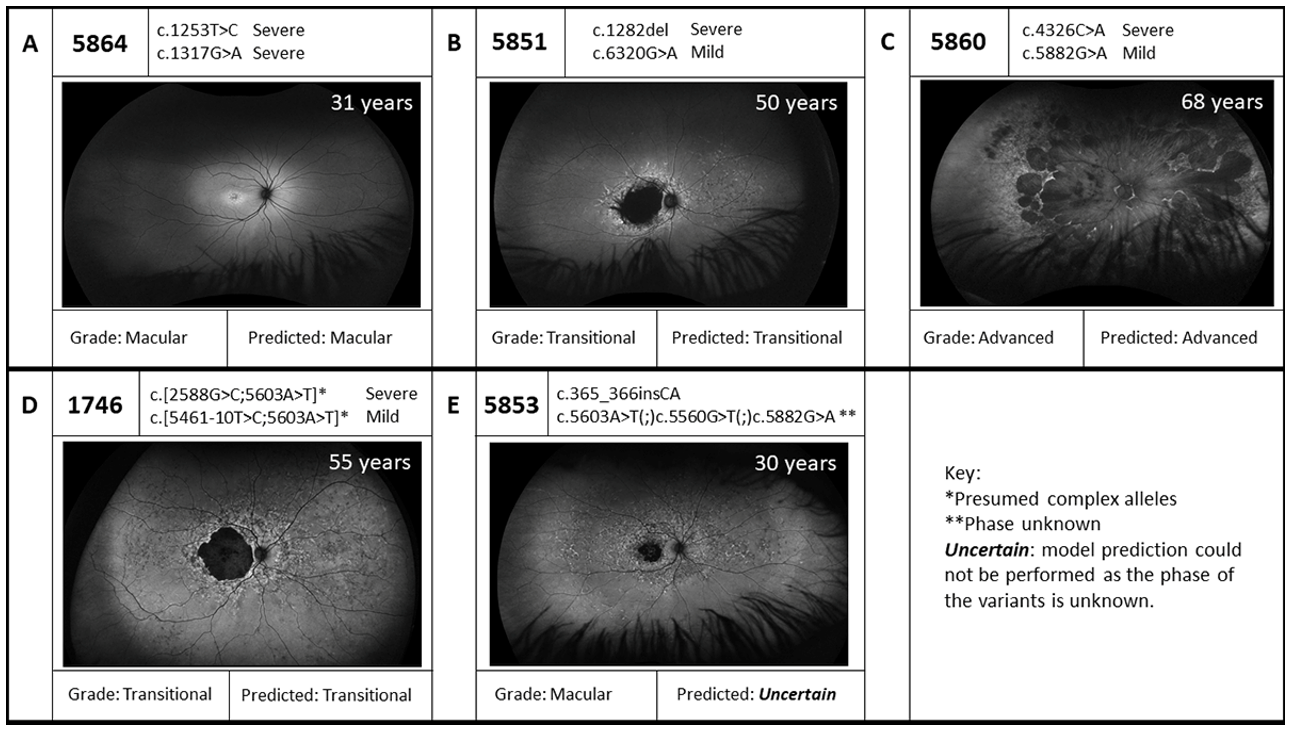
| 1337 | 59 | 3 | STGD | p.(Asn1868Ile)(;)(Leu1940Pro) | MildLP Severe | p.(?) † | Severe | STGD | Uncertain |
| 1746 | 55 | 3 | STGD | p.[[Gly863Ala,Gly863del]; (Asn1868Ile)] | Mild | p.[[Thr1821Aspfs*6,Thr1821Valfs*13]; (Asn1868Ile)] | Severe | STGD | Yes |
| 1808 | 34 | 1 | STGD | p.[[Thr1821Aspfs*6,Thr1821Valfs*13];(Asn1868Ile)] | Severe | p.(Gly1961Glu) | Mild | STGD | Yes |
| 2469 | 36 | 3 | STGD | p.[[Gly863Ala,Gly863del]; (Asn1868Ile)] | Mild | p.[[Thr1821Aspfs*6,Thr1821Valfs*13]; (Asn1868Ile)] | Severe | STGD | Yes |
| 2843 | 22 | 3 | CRD | p.(Cys1339Tyr)(;)(?) † | Unknown Severe | p.(Arg2030*) | Severe | STGD | Uncertain |
| 3536 | 60 | 1/2 | Late onset STGD | p.(Ala1038Val) | Mild | p.(Gln636*) | Severe | STGD | No |
| 4126 | 13 | 1/2 | STGD | p.[=;Gly1592Alafs*113](;) [=;Met1733Glufs*78] | Benign Mild | p.[[Thr1821Aspfs*6,Thr1821Valfs*13]; (Asn1868Ile)] | Severe | STGD | Uncertain |
| 5219 | 68 | 4 | Early onset STGD | p.(Arg212Cys) | Severe | p.[[Thr1821Aspfs*6,Thr1821Valfs*13]; (Asn1868Ile)] | Severe | Early onset STGD | Yes |
| 5349 | 40 | 2 | STGD | p.(Gln636*) | Severe | p.(Asn1868Ile) | MildLP | STGD | Yes |
| 5604 | 54 | 4 | STGD | p.(Thr1526Met)(;)(Cys1490Tyr) | Moderate Severe | p.(Asn1868Ile) | MildLP | STGD | Uncertain |
| 5607 | 33 | 2 | STGD | p.(Glu1087Lys) | Severe | p.(Arg2030Gln) | Mild | STGD | Yes |
| 5608 | 45 | 3 | STGD | p.[[Gly863Ala,Gly863del]; (Asn1868Ile)] | Mild | p.[[Thr1821Aspfs*6,Thr1821Valfs*13]; (Asn1868Ile)] | Severe | STGD | Yes |
| 5609 | 37 | 4 | STGD | p.(Pro1380Leu) | Moderate | p.(Pro1380Leu) | Moderate | STGD | No |
| 5851 | 50 | 3 | STGD | p.(Val428Serfs*7) | Severe | p.(Arg2107His) | Mild | STGD | Yes |
| 5852 | 66 | 3 | STGD | p.[[Gly863Ala,Gly863del]; (Asn1868Ile)] | Mild | p.[[Thr1821Aspfs*6,Thr1821Valfs*13]; (Asn1868Ile)] | Severe | STGD | Yes |
| 5853 | 30 | 1 | STGD | p.(Gly123Metfs*32) | Severe | p.(Val1854Leu)(;)(Asn1868Ile)(;) (Gly1961Glu) | Severe MildLP Mild | STGD | Uncertain |
| 5854 | 36 | 2 | STGD | p.(Glu1399Lys) | Mild | p.(Ala1773Val) | Severe | STGD | Yes |
| 5857 | 20 | 3 | Early onset STGD | p.(Arg2077Trp) | Severe | p.(Arg2077Trp) | Severe | Early onset STGD | Yes |
| 5860 | 31 | 1 | STGD | p.(Asn1442Lys) | Severe | p.(Gly1961Glu) | Mild | STGD | Yes |
| 5861 | 37 | 3 | STGD | p.[=,Glu1863Leufs*33] | Moderate | p.[[Thr1821Aspfs*6,Thr1821Valfs*13]; (Asn1868Ile)] | Severe | STGD | Yes |
| 5862 | 17 | 3 | Early onset STGD | p.(Gln636*) | Severe | p.(Thr1526Met) | Moderate | Early onset STGD | Yes |
| 5863 | 42 | 2 | STGD | p.[[Gly863Ala,Gly863del]; (Asn1868Ile)] | Mild | p.(Gln1513Argfs*13) | Severe | STGD | Yes |
| 5864 | 68 | 4 | STGD | p.(Phe418Ser) | Severe | p.(Trp439*) | Severe | STGD | Yes |
| 5865 | 49 | 2 | STGD | p.(Gln636*) | Severe | p.(Asn1868Ile) | MildLP | STGD | Yes |
| 3656 | 42 | 1 | Occult MD | p.(Pro1380Leu) | Moderate | p.(Gly1961Glu) | Mild | STGD | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mc Clinton, B.; Corradi, Z.; McKibbin, M.; Panneman, D.M.; Roosing, S.; Boonen, E.G.M.; Ali, M.; Watson, C.M.; Steel, D.H.; Cremers, F.P.M.; et al. Effective smMIPs-Based Sequencing of Maculopathy-Associated Genes in Stargardt Disease Cases and Allied Maculopathies from the UK. Genes 2023, 14, 191. https://doi.org/10.3390/genes14010191
Mc Clinton B, Corradi Z, McKibbin M, Panneman DM, Roosing S, Boonen EGM, Ali M, Watson CM, Steel DH, Cremers FPM, et al. Effective smMIPs-Based Sequencing of Maculopathy-Associated Genes in Stargardt Disease Cases and Allied Maculopathies from the UK. Genes. 2023; 14(1):191. https://doi.org/10.3390/genes14010191
Chicago/Turabian StyleMc Clinton, Benjamin, Zelia Corradi, Martin McKibbin, Daan M. Panneman, Susanne Roosing, Erica G. M. Boonen, Manir Ali, Christopher M. Watson, David H. Steel, Frans P. M. Cremers, and et al. 2023. "Effective smMIPs-Based Sequencing of Maculopathy-Associated Genes in Stargardt Disease Cases and Allied Maculopathies from the UK" Genes 14, no. 1: 191. https://doi.org/10.3390/genes14010191
APA StyleMc Clinton, B., Corradi, Z., McKibbin, M., Panneman, D. M., Roosing, S., Boonen, E. G. M., Ali, M., Watson, C. M., Steel, D. H., Cremers, F. P. M., Inglehearn, C. F., Hitti-Malin, R. J., & Toomes, C. (2023). Effective smMIPs-Based Sequencing of Maculopathy-Associated Genes in Stargardt Disease Cases and Allied Maculopathies from the UK. Genes, 14(1), 191. https://doi.org/10.3390/genes14010191