
Comparative Analysis of Chloroplast Genomes within Saxifraga (Saxifragaceae) Takes Insights into Their Genomic Evolution and Adaption to the High-Elevation Environment

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material Sampling and DNA Sequencing
2.2. De Novo Assembly and Annotation
2.3. Sequence Divergence Analysis and Visualization
2.4. Calculation of Codon Usage
2.5. Identification of Repeat Sequences in Organelle Genomes
2.6. Phylogenetic Analysis
2.7. Selective Analysis
3. Results and Discussion
3.1. Characteristics of the CP Genome for Saxifraga Species
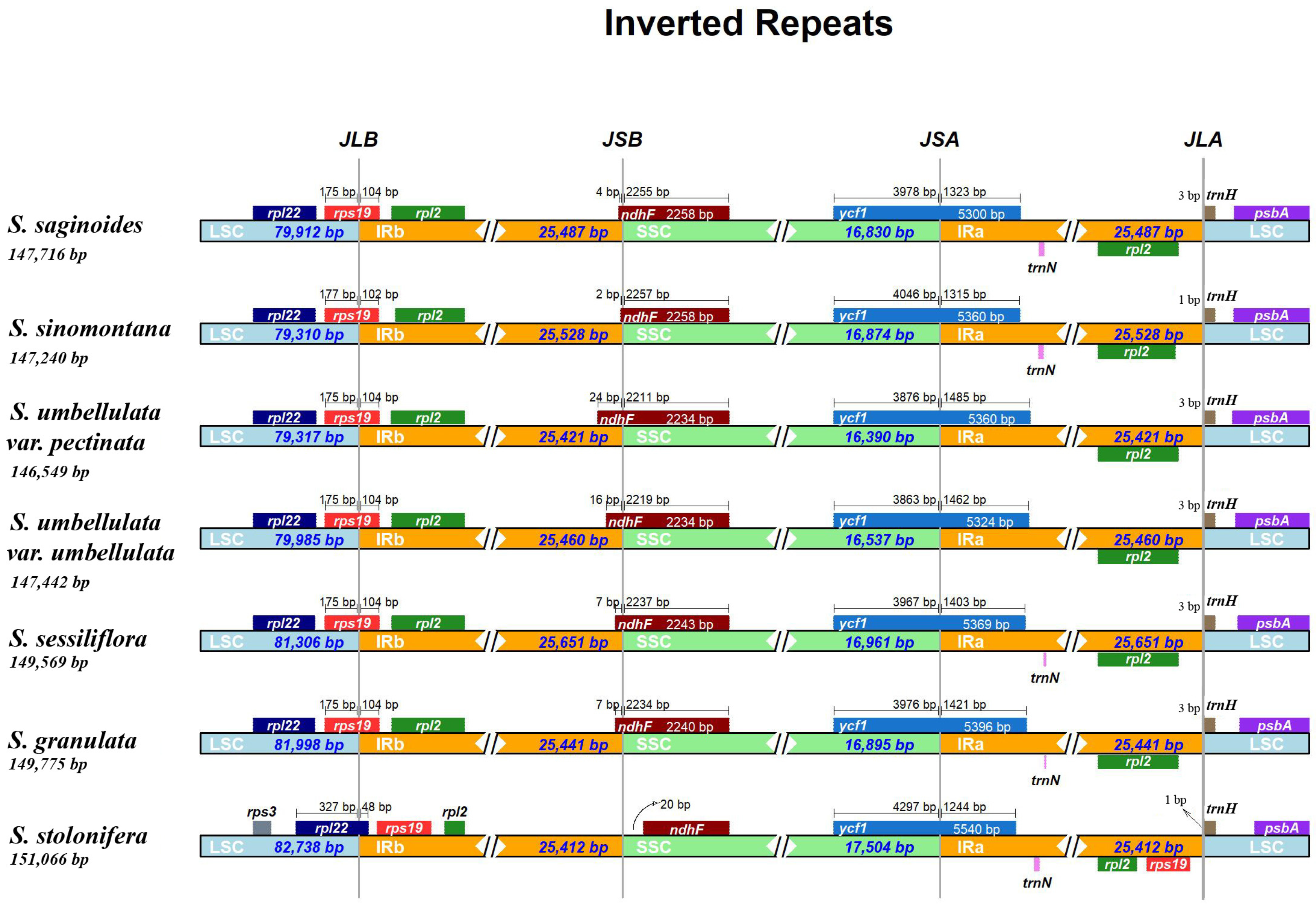
3.2. IR Boundary Analysis
3.3. Genomic Sequence Divergence
3.4. Condon Usage Analysis
3.5. Repeat Sequence Analysis
3.6. Phylogenetic Analysis
3.7. Selection and Adaption Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moreira, D.; Le Guyader, H.; Philippe, H. The origin of red algae and the evolution of chloroplasts. Nature 2000, 405, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.; Khurana, J.; Tyagi, A.; Khurana, P. An update on chloroplast genomes. Plant Syst. Evol. 2008, 271, 101–122. [Google Scholar] [CrossRef]
- Jansen, R.K.; Ruhlman, T.A. Plastid genomes of seed plants. In Genomics of Chloroplasts and Mitochondria; Springer: Berlin/Heidelberg, Germany, 2012; pp. 103–126. [Google Scholar]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef]
- Sloan, D.B.; Triant, D.A.; Forrester, N.J.; Bergner, L.M.; Wu, M.; Taylor, D.R. A recurring syndrome of accelerated plastid genome evolution in the angiosperm tribe Sileneae (Caryophyllaceae). Mol. Phylogenetics Evol. 2014, 72, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.L.; Wang, R.N.; Zhang, N.Y.; Fan, W.B.; Fang, M.F.; Li, Z.H. Molecular Evolution of Chloroplast Genomes of Orchid Species: Insights into Phylogenetic Relationship and Adaptive Evolution. Int. J. Mol. Sci. 2018, 19, 716. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.-F.; Zhang, Y.-X.; Zeng, C.-X.; Guo, Z.-H.; Li, D.-Z. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 2014, 63, 933–950. [Google Scholar] [CrossRef]
- Mehmood, F.; Ubaid, Z.; Bao, Y.; Poczai, P.; Mirza, B. Comparative plastomics of Ashwagandha (Withania, Solanaceae) and identification of mutational hotspots for barcoding medicinal plants. Plants 2020, 9, 752. [Google Scholar] [CrossRef]
- Flora of China. Available online: https://www.iplant.cn/foc (accessed on 10 March 2020).
- Zhmylev, P. Rod Saxifraga L. (Saxifragaceae): Biomorfologia, Sistematika i Evolutsia Zhiznennykh Form [Genus Saxifraga L. (Saxifragaceae): Biomorphology, Systematics and Evolution of the Life Forms]. Ph.D. Thesis, Moscow State University, Moscow, Russia, 2004. [Google Scholar]
- Mas De Xaxars, G.; García-Fernández, A.; Barnola, P.; Martín, J.; Mercadé, A.; Vallès, J.; Vargas, P.; Vigo, J.; Garnatje, T. Phylogenetic and cytogenetic studies reveal hybrid speciation in Saxifraga subsect. Triplinervium (Saxifragaceae). J. Syst. Evol. 2015, 53, 53–62. [Google Scholar] [CrossRef]
- Badral, D.; Odonbayar, B.; Murata, T.; Munkhjargal, T.; Tuvshintulga, B.; Igarashi, I.; Suganuma, K.; Inoue, N.; Brantner, A.H.; Odontuya, G. Flavonoid and galloyl glycosides isolated from Saxifraga spinulosa and their antioxidative and inhibitory activities against species that cause piroplasmosis. J. Nat. Prod. 2017, 80, 2416–2423. [Google Scholar] [CrossRef]
- Dang, J.; Tao, Y.; Shao, Y.; Mei, L.; Zhang, L.; Wang, Q. Antioxidative extracts and phenols isolated from Qinghai–Tibet Plateau medicinal plant Saxifraga tangutica Engl. Ind. Crops Prod. 2015, 78, 13–18. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, Y.-M.; Yang, S.; Song, B.-A.; Xu, G.-F.; Bhadury, P.S.; Jin, L.-H.; Hu, D.-Y.; Liu, F.; Xue, W. Studies on the chemical constituents and anticancer activity of Saxifraga stolonifera (L) Meeb. Bioorg Med. Chem. 2008, 16, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Ebersbach, J.; Muellner-Riehl, A.; Michalak, I.; Tkach, N.; Hoffmann, M.; Röser, M.; Sun, H.; Favre, A. In and out of the Qinghai-Tibet Plateau: Divergence time estimation and historical biogeography of the large arctic-alpine genus Saxifraga L. J. Biogeogr. 2017, 44, 900–910. [Google Scholar] [CrossRef]
- Ebersbach, J.; Schnitzler, J.; Favre, A.; Muellner-Riehl, A. Evolutionary radiations in the species-rich mountain genus Saxifraga L. BMC Evol. Biol. 2017, 17, 119. [Google Scholar] [CrossRef] [PubMed]
- Conti, E.; Soltis, D.E.; Hardig, T.M.; Schneider, J. Phylogenetic relationships of the silver saxifrages (Saxifraga, sect. Ligulatae Haworth): Implications for the evolution of substrate specificity, life histories, and biogeography. Mol. Phylogenetics Evol. 1999, 13, 536–555. [Google Scholar] [CrossRef] [PubMed]
- Guldahl, A.S.; Gabrielsen, T.M.; Scheen, A.-C.; Borgen, L.; Steen, S.W.; Spjelkavik, S.; Brochmann, C. The Saxifraga rivularis complex in Svalbard: Molecules, ploidy and morphology. Flora-Morphol. Distrib. Funct. Ecol. Plants 2005, 200, 207–221. [Google Scholar] [CrossRef]
- Gao, Q.-B.; Li, Y.-H.; Gornall, R.J.; Zhang, Z.-X.; Zhang, F.-Q.; Xing, R.; Fu, P.-C.; Wang, J.-L.; Liu, H.-R.; Tian, Z.-Z. Phylogeny and speciation in Saxifraga sect. Ciliatae (Saxifragaceae): Evidence from psbA-trnH, trnL-F and ITS sequences. Taxon 2015, 64, 703–713. [Google Scholar] [CrossRef]
- Vargas, P. A phylogenetic study of Saxifraga sect. Saxifraga (Saxifragaceae) based on nrDNA ITS sequences. Plant Syst. Evol. 2000, 223, 59–70. [Google Scholar] [CrossRef]
- Tkach, N.; Röser, M.; Miehe, G.; Muellner-Riehl, A.N.; Ebersbach, J.; Favre, A.; Hoffmann, M.H. Molecular phylogenetics, morphology and a revised classification of the complex genus Saxifraga (Saxifragaceae). Taxon 2015, 64, 1159–1187. [Google Scholar] [CrossRef]
- Wang, J.; Wang, M.; Zhang, X.; Sun, S.; Chen, N.; Zhang, A.; Zhao, C. Picea purpurea has a physiological advantage over its progenitors in alpine ecosystems due to transgressive segregation. J. For. Res. 2018, 23, 363–371. [Google Scholar] [CrossRef]
- Peng, Y.; Yang, Z.; Zhang, H.; Cui, C.; Qi, X.; Luo, X.; Tao, X.; Wu, T.; Ouzhuluobu; Basang; et al. Genetic variations in Tibetan populations and high-altitude adaptation at the Himalayas. Mol. Biol. Evol. 2011, 28, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Casati, P.; Stapleton, A.E.; Blum, J.E.; Walbot, V. Genome-wide analysis of high-altitude maize and gene knockdown stocks implicates chromatin remodeling proteins in response to UV-B. Plant J. 2006, 46, 613–627. [Google Scholar] [CrossRef]
- Guo, X.; Hu, Q.; Hao, G.; Wang, X.; Zhang, D.; Ma, T.; Liu, J. The genomes of two Eutrema species provide insight into plant adaptation to high altitudes. DNA Res. 2018, 25, 307–315. [Google Scholar] [CrossRef]
- Zhang, J.; Tian, Y.; Yan, L.; Zhang, G.; Wang, X.; Zeng, Y.; Zhang, J.; Ma, X.; Tan, Y.; Long, N.; et al. Genome of Plant Maca (Lepidium meyenii) Illuminates Genomic Basis for High-Altitude Adaptation in the Central Andes. Mol. Plant 2016, 9, 1066–1077. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, X.; Landis, J.B.; Zhang, H.; Deng, T.; Sun, H.; Wang, H. Plastome evolution in Dolomiaea (Asteraceae, Cardueae) using phylogenomic and comparative analyses. Front. Plant Sci. 2020, 11, 376. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.-N.; Ren, Y.; Zhang, J.-Q. Conservation and innovation: Plastome evolution during rapid radiation of Rhodiola on the Qinghai-Tibetan Plateau. Mol. Phylogenetics Evol. 2020, 144, 106713. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; DePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 1–31. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-Y.-F. figshare. Available online: https://doi.org/10.6084/m9.figshare.20488218.v2 (accessed on 19 August 2022).
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Brudno, M.; Do, C.B.; Cooper, G.M.; Kim, M.F.; Davydov, E.; Green, E.D.; Sidow, A.; Batzoglou, S.; Program, N.C.S. LAGAN and Multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003, 13, 721–731. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Wei, L.; He, J.; Jia, X.; Qi, Q.; Liang, Z.; Zheng, H.; Ping, Y.; Liu, S.; Sun, J. Analysis of codon usage bias of mitochondrial genome in Bombyx moriand its relation to evolution. BMC Evol. Biol. 2014, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Rasool, S.T. Analysis of synonymous codon usage in Zika virus. Acta Trop. 2017, 173, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Zhang, Z.; Xiao, J.; Wu, J.; Zhang, H.; Liu, G.; Wang, X.; Dai, L. ParaAT: A parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 2012, 419, 779–781. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating γ-series methods and sliding window strategies. Genom Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Wu, Z.; Liao, R.; Yang, T.; Dong, X.; Lan, D.; Qin, R.; Liu, H. Analysis of six chloroplast genomes provides insight into the evolution of Chrysosplenium (Saxifragaceae). BMC Genom. 2020, 21, 621. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Xu, Q.; Liu, K.; Zhao, Y.; Chen, N. Chloroplast genomes for five Skeletonema species: Comparative and phylogenetic analysis. Front. Plant Sci. 2021, 12, 774617. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Yu, X.; Zhou, J.; Li, J.; Tian, Z.; Wang, P.; Meng, Y.; Zhao, Q.; Lou, Q.; Du, S. Complete chloroplast genome sequencing and comparative analysis reveals changes to the chloroplast genome after allopolyploidization in Cucumis. Genome 2021, 64, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-L.; Jansen, R.K.; Chumley, T.W.; Kim, K.-J. Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Mol. Biol. Evol. 2007, 24, 1161–1180. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef]
- Menezes, A.P.A.; Resende-Moreira, L.C.; Buzatti, R.S.O.; Nazareno, A.G.; Carlsen, M.; Lobo, F.P.; Kalapothakis, E.; Lovato, M.B. Chloroplast genomes of Byrsonima species (Malpighiaceae): Comparative analysis and screening of high divergence sequences. Sci. Rep. 2018, 8, 2210. [Google Scholar] [CrossRef]
- Nock, C.J.; Waters, D.L.; Edwards, M.A.; Bowen, S.G.; Rice, N.; Cordeiro, G.M.; Henry, R.J. Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 2011, 9, 328–333. [Google Scholar] [CrossRef]
- Dong, W.; Liu, H.; Xu, C.; Zuo, Y.; Chen, Z.; Zhou, S. A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: A case study on ginsengs. BMC Genet. 2014, 15, 138. [Google Scholar] [CrossRef]
- A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [CrossRef]
- Xu, C.; Cai, X.; Chen, Q.; Zhou, H.; Cai, Y.; Ben, A. Factors affecting synonymous codon usage bias in chloroplast genome of oncidium gower ramsey. Evol. Bioinform. 2011, 7, 271–278. [Google Scholar] [CrossRef]
- Das, S.; Paul, S.; Dutta, C. Synonymous codon usage in adenoviruses: Influence of mutation, selection and protein hydropathy. Virus Res. 2006, 117, 227–236. [Google Scholar] [CrossRef]
- McClellan, D.A. The codon-degeneracy model of molecular evolution. J. Mol. Evol. 2000, 50, 131–140. [Google Scholar] [CrossRef]
- Zhang, P.; Xu, W.; Lu, X.; Wang, L. Analysis of codon usage bias of chloroplast genomes in Gynostemma species. Physiol. Mol. Biol. Plants 2021, 27, 2727–2737. [Google Scholar] [CrossRef]
- Tuler, A.C.; Carrijo, T.T.; Nóia, L.R.; Ferreira, A.; Peixoto, A.L.; da Silva Ferreira, M.F. SSR markers: A tool for species identification in Psidium (Myrtaceae). Mol. Biol. Rep. 2015, 42, 1501–1513. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.H.; Zhang, J.J.; Yao, X.H.; Huang, H.W. Chloroplast microsatellite markers in Liriodendron tulipifera (Magnoliaceae) and cross-species amplification in L. chinense. Am. J. Bot. 2011, 98, e123–e126. [Google Scholar] [CrossRef] [PubMed]
- Korkovelos, A.; Mavromatis, A.; Huang, W.; Hagidimitriou, M.; Giakoundis, A.; Goulas, C. Effectiveness of SSR molecular markers in evaluating the phylogenetic relationships among eight Actinidia species. Sci. Hortic. 2008, 116, 305–310. [Google Scholar] [CrossRef]
- Tabbasam, N.; Zafar, Y. Pros and cons of using genomic SSRs and EST-SSRs for resolving phylogeny of the genus Gossypium. Plant Syst. Evol. 2014, 300, 559–575. [Google Scholar] [CrossRef][Green Version]
- Cavalier-Smith, T. Chloroplast evolution: Secondary symbiogenesis and multiple losses. Curr. Biol. 2002, 12, R62–R64. [Google Scholar] [CrossRef]
- Powell, W.; Morgante, M.; McDevitt, R.; Vendramin, G.G.; Rafalski, J.A. Polymorphic simple sequence repeat regions in chloroplast genomes: Applications to the population genetics of pines. Proc. Natl. Acad. Sci. USA 1995, 92, 7759–7763. [Google Scholar] [CrossRef]
- Liu, W.; Kong, H.; Zhou, J.; Fritsch, P.W.; Hao, G.; Gong, W. Complete Chloroplast Genome of Cercis chuniana (Fabaceae) with Structural and Genetic Comparison to Six Species in Caesalpinioideae. Int. J. Mol. Sci. 2018, 19, 1286. [Google Scholar] [CrossRef]
- Deng, J.B.; Drew, B.T.; Mavrodiev, E.V.; Gitzendanner, M.A.; Soltis, P.S.; Soltis, D.E. Phylogeny, divergence times, and historical biogeography of the angiosperm family Saxifragaceae. Mol. Phylogenetics Evol. 2015, 83, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.X.; Du, Y.X.; Folk, R.A.; Wang, S.Y.; Soltis, D.E.; Shang, F.D.; Li, P. Plastome Evolution in Saxifragaceae and Multiple Plastid Capture Events Involving Heuchera and Tiarella. Front. Plant Sci. 2020, 11, 361. [Google Scholar] [CrossRef] [PubMed]
- Sirpiö, S.; Allahverdiyeva, Y.; Holmström, M.; Khrouchtchova, A.; Haldrup, A.; Battchikova, N.; Aro, E.M. Novel nuclear-encoded subunits of the chloroplast NAD(P)H dehydrogenase complex. J. Biol. Chem. 2009, 284, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Kamruzzaman Munshi, M.; Kobayashi, Y.; Shikanai, T. Identification of a novel protein, CRR7, required for the stabilization of the chloroplast NAD(P)H dehydrogenase complex in Arabidopsis. Plant J. 2005, 44, 1036–1044. [Google Scholar] [CrossRef]
- Wang, P.; Duan, W.; Takabayashi, A.; Endo, T.; Shikanai, T.; Ye, J.Y.; Mi, H. Chloroplastic NAD(P)H dehydrogenase in tobacco leaves functions in alleviation of oxidative damage caused by temperature stress. Plant Physiol. 2006, 141, 465–474. [Google Scholar] [CrossRef] [PubMed]
- De Vries, J.; Sousa, F.L.; Bölter, B.; Soll, J.; Gould, S.B. YCF1: A green TIC? Plant Cell 2015, 27, 1827–1833. [Google Scholar] [CrossRef]
- Rochaix, J.D. Chloroplast protein import machinery and quality control. FEBS J. 2022. [Google Scholar] [CrossRef]
- Bölter, B.; Soll, J. Ycf1/Tic214 Is Not Essential for the Accumulation of Plastid Proteins. Mol. Plant 2017, 10, 219–221. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Group | Genes |
|---|---|---|
| Photosynthesis related genes | Rubisco | rbcL |
| Photosystem I | psaA, psaB, psaC, psaI, psaJ | |
| Photosystem II | psbA, psbB, psbT, psbK, psbI, psbH, psbM, psbN, psbD, psbC, psbZ, psbJ, psbL, psbE, psbF | |
| ATP synthase | atpA, atpB, atpE, atpF a, atpH, atpI | |
| Cytochrome b/f complex | petA, petB a, petD, petN, petL, petG | |
| Cytochrome C synthesis | ccsA | |
| NADPH dehydrogenase | NdhA a, ndhB a,c (×2), ndhC, ndhD, ndhE, ndhF, ndhH, ndhG, ndhJ, ndhK, ndhI | |
| Transcription and translation related genes | Transcription | rpoA, rpoB, rpoC2, rpoC1 a |
| Ribosomal proteins | rps2, rps3, rps4, rps7 c (×2), rps8, rps11, rps12 b,c (×2), rps14, rps15, rps16 a, rps18, rps19, rpl2 a,c (×2), rpl14, rpl16 a, rpl20, rpl22, rpl23 c (×2), rpl32, rpl33, rpl36 | |
| Translation initiation factor | infA | |
| RNA genes | Ribosomal RNA | rrn16S c (×2), rrn23S c (×2), rrn4.5 c (×2), rrn5 c (×2) |
| Transfer RNA | trnH-GUG, trnK-UUU a, trnQ-UUG, trnS-GCU, trnS-UGA, trnS-GGA, trnG-GCC a, trnR-UCU, trnR-ACG c (×2), trnC-GCA, trnD-GUC, trnY-GUA, trnE-UUC, trnT-UGU, trnG-UCC, trnfM-CAU, trnL-CAA c (×2), trnL-UAA a, trnL-UAG, trnF-GAA, trnV-GAC c (×2), trnV-UAC a, trnM-CAU, trnT-GGU, trnW-CCA, trnP-UGG, trnI-CAU c (×2), trnI-GAU a,c (×2), trnA-UGC a,c (×2), trnN-GUU c (×2) | |
| Other genes | RNA processing | matK |
| Carbon metabolism | cemA | |
| Fatty acid synthesis | accD | |
| Proteolysis | ClpP b | |
| Conserved ORFs | ycf1, ycf2 c (×2), ycf3 b, ycf4 |
| Genome Feature | S. saginoides | S. sessiliflora | S. sinomontana | S. umbellulata var. pectinata | S. umbellulata var. umbellulata | S. granulata | S. stolonifera |
|---|---|---|---|---|---|---|---|
| Genome size (bp) | 147,716 | 149,569 | 147,240 | 146,549 | 147,442 | 149,775 | 151,066 |
| LSC size (bp) | 79,912 | 81,306 | 79,310 | 79,317 | 79,985 | 81,998 | 82,738 |
| SSC size (bp) | 16,830 | 16,961 | 16,874 | 16,390 | 16,537 | 16,895 | 17,504 |
| IR size (bp) | 25,487 | 25,651 | 25,528 | 25,421 | 25,460 | 25,441 | 25,412 |
| Number of genes | 130 | 130 | 130 | 130 | 130 | 130 | 131 |
| Protein genes | 85 | 85 | 85 | 85 | 85 | 85 | 86 |
| tRNA genes | 37 | 37 | 37 | 37 | 37 | 37 | 37 |
| rRNA genes | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| Duplicated genes in IRs | 17 | 17 | 17 | 17 | 17 | 17 | 18 |
| GC content (%) | 38% | 37.9% | 38% | 38.1% | 38.1% | 37.8% | 37.8% |
| GC content in LSC (%) | 36.2% | 36% | 36.2% | 36.2% | 36.2% | 35.8% | 35.9% |
| GC content in SSC (%) | 32.1% | 32% | 32% | 32.4% | 32.4% | 31.9% | 32.2% |
| GC content in IRs (%) | 42.9% | 42.8% | 42.9% | 42.8% | 42.8% | 42.9% | 43% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.; Yu, X.; Yang, Y.; Wei, P.; Zhang, W.; Li, X.; Liu, C.; Zhao, S.; Li, X.; Liu, X. Comparative Analysis of Chloroplast Genomes within Saxifraga (Saxifragaceae) Takes Insights into Their Genomic Evolution and Adaption to the High-Elevation Environment. Genes 2022, 13, 1673. https://doi.org/10.3390/genes13091673
Chen Z, Yu X, Yang Y, Wei P, Zhang W, Li X, Liu C, Zhao S, Li X, Liu X. Comparative Analysis of Chloroplast Genomes within Saxifraga (Saxifragaceae) Takes Insights into Their Genomic Evolution and Adaption to the High-Elevation Environment. Genes. 2022; 13(9):1673. https://doi.org/10.3390/genes13091673
Chicago/Turabian StyleChen, Zhuyifu, Xiaolei Yu, Yujiao Yang, Pei Wei, Wencai Zhang, Xinzhong Li, Chenlai Liu, Shuqi Zhao, Xiaoyan Li, and Xing Liu. 2022. "Comparative Analysis of Chloroplast Genomes within Saxifraga (Saxifragaceae) Takes Insights into Their Genomic Evolution and Adaption to the High-Elevation Environment" Genes 13, no. 9: 1673. https://doi.org/10.3390/genes13091673
APA StyleChen, Z., Yu, X., Yang, Y., Wei, P., Zhang, W., Li, X., Liu, C., Zhao, S., Li, X., & Liu, X. (2022). Comparative Analysis of Chloroplast Genomes within Saxifraga (Saxifragaceae) Takes Insights into Their Genomic Evolution and Adaption to the High-Elevation Environment. Genes, 13(9), 1673. https://doi.org/10.3390/genes13091673