Comparative Transcriptome Analyses of Different Rheum officinale Tissues Reveal Differentially Expressed Genes Associated with Anthraquinone, Catechin, and Gallic Acid Biosynthesis

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction, cDNA Library Construction, and Illumina Sequencing
2.3. De Novo Assembly and Functional Annotation
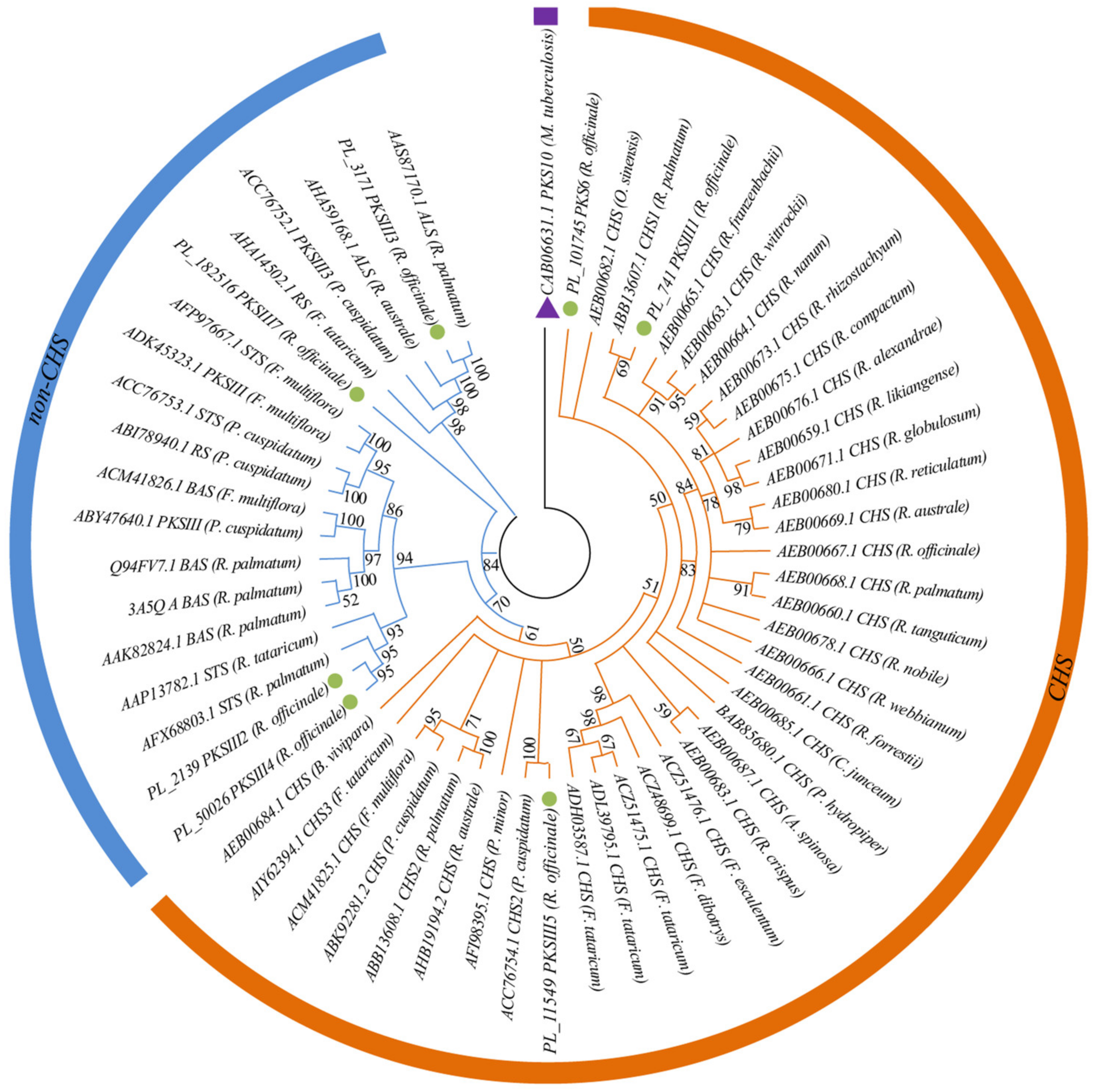
2.4. Phylogenetic Analyses of Type III Polyketide Synthases (PKS III) Genes in R. officinale
2.5. Differential Gene Expression Analyses
2.6. Transcription Factor Analysis
2.7. Quantitative Real-Time PCR Verification
2.8. Identification of Simple Sequence Repeat (SSR)
3. Results
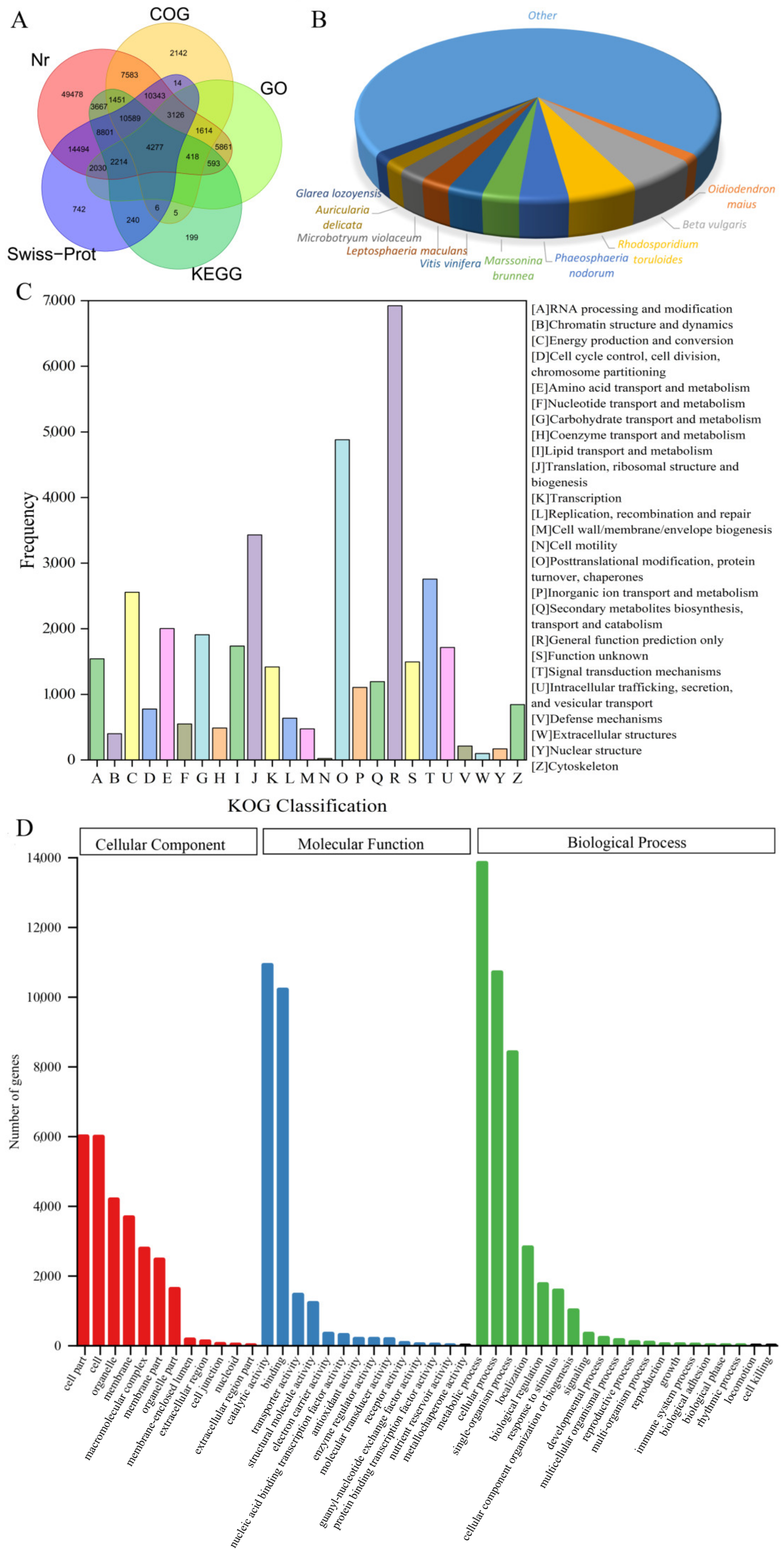
3.1. Subsection RNA Sequencing and De Novo Assembly
3.2. Functional Annotation
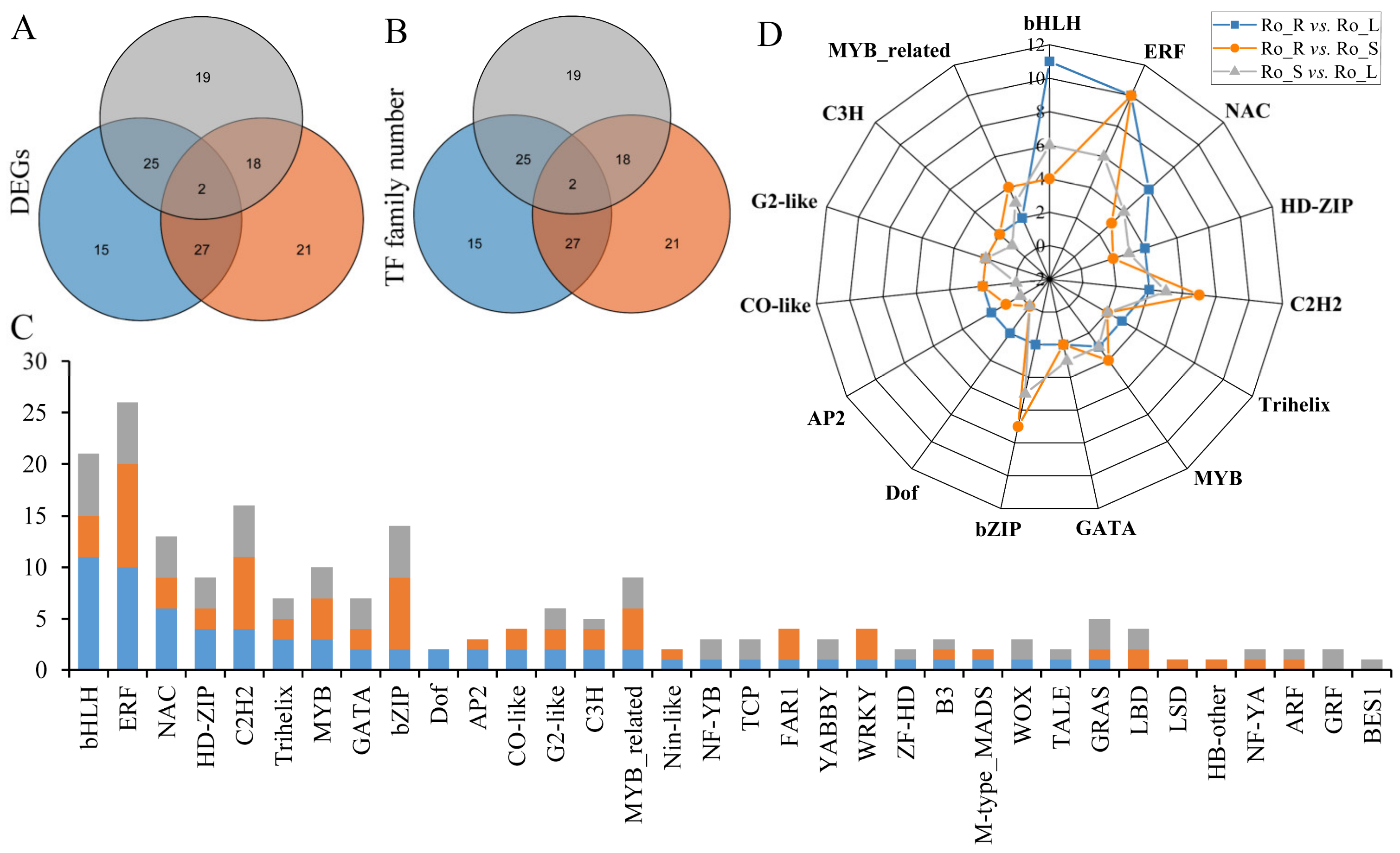
3.3. Differentially Expressed Gene Analyses between Different Tissues
3.4. Transcription Factors (TFs) Analysis of DEGs
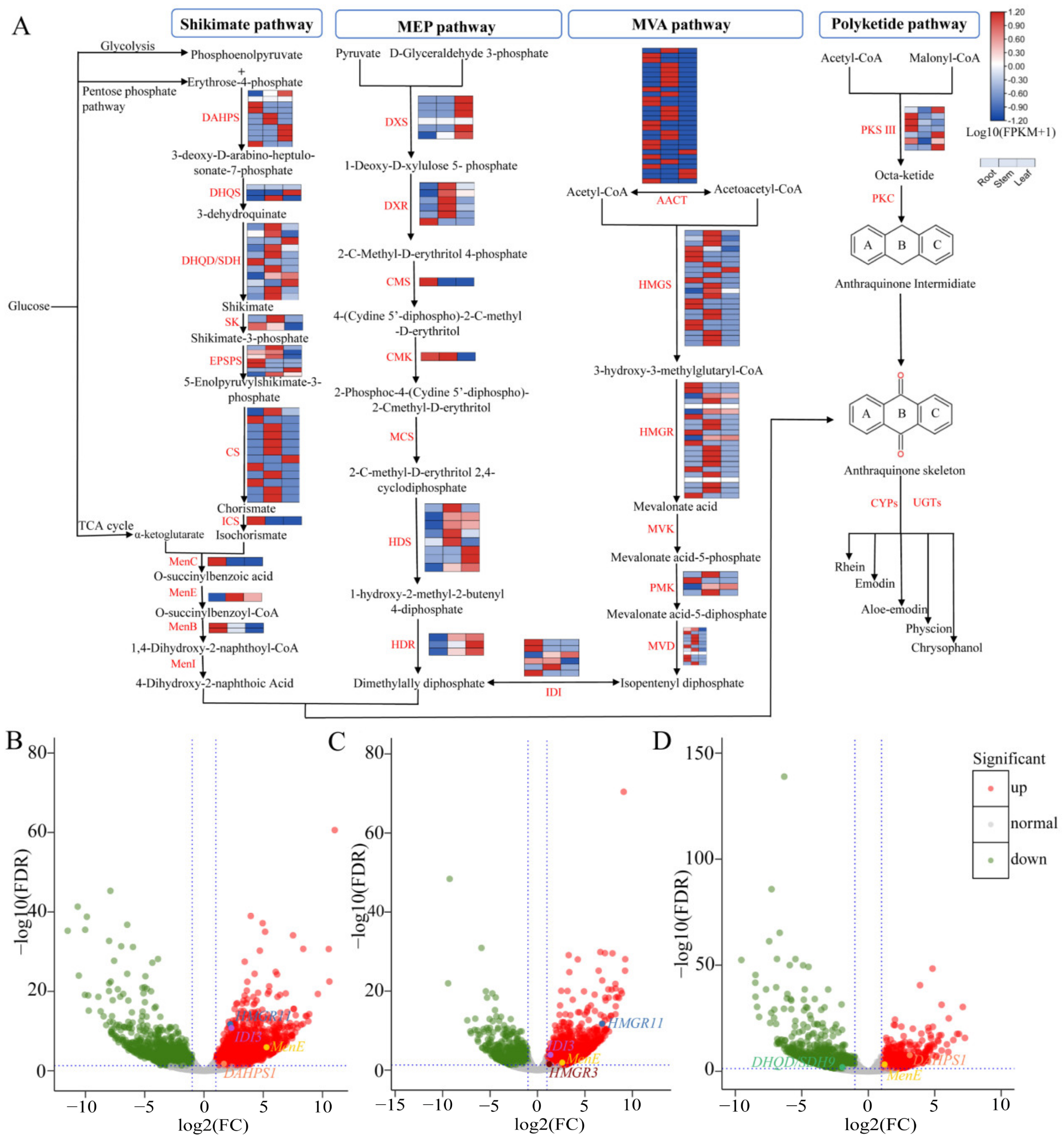
3.5. Identification of Genes Associated with the Anthraquinone Biosynthesis
3.6. Catechin and Gallic Acid Biosynthesis in R. officinale
3.7. Quantitative Real-Time PCR (qRT-PCR) Validation
3.8. Detection of SSR Loci
4. Discussion
4.1. De Novo Assembly and Functional Annotation
4.2. Candidate Genes Identification and DEG Analyses Associated with Secondary Metabolite Biosynthesis
4.3. Transcription Factor Analysis
4.4. Simple Sequence Repeat (SSR) Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Yang, W.T.; Ke, C.Y.; Wu, W.T.; Tseng, Y.H.; Lee, R.P. Antimicrobial and anti-inflammatory potential of Angelica dahurica and Rheum officinale extract accelerates wound healing in Staphylococcus aureus-infected wounds. Sci. Rep. 2020, 10, 5596. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Li, J.; He, M.; Ouyang, H.; Ruan, L.; Huang, X.; Rao, Y.; Yang, S.; Zhou, X.; Bai, J. Investigation and identification of the multiple components of Rheum officinale Baill. using ultra-high-performance liquid chromatography coupled with quadrupole-time-of-flight tandem mass spectrometry and data mining strategy. J. Sep. Sci. 2021, 44, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.J.; Pu, Z.J.; Tang, Y.P.; Shen, J.; Chen, Y.Y.; Kang, A.; Zhou, G.S.; Duan, J.A. Advances in bio-active constituents, pharmacology and clinical applications of rhubarb. Chin. Med. 2017, 12, 36. [Google Scholar] [CrossRef]
- Chao, Y.H.; Yang, W.T.; Li, M.C.; Yang, F.L.; Lee, R.P. Angelica dahurica and Rheum officinale facilitated diabetic wound healing by elevating vascular endothelial growth factor. Am. J. Chin. Med. 2021, 49, 1515–1533. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Dai, Z.; Chen, X.; Xie, D.; Tang, Q.; Cheng, C.; Xu, Y.; Deng, C.; Liu, C.; Chen, J.; et al. Gene coexpression network analysis and tissue-specific profiling of gene expression in jute (Corchorus capsularis L.). BMC Genom. 2020, 21, 406. [Google Scholar] [CrossRef]
- Rolta, R.; Salaria, D.; Sharma, P.; Sharma, B.; Kumar, V.; Rathi, B.; Verma, M.; Sourirajan, A.; Baumler, D.J.; Dev, K. Phytocompounds of Rheum emodi, Thymus serpyllum, and Artemisia annua inhibit spike protein of SARS-CoV-2 binding to ACE2 receptor: In Silico approach. Curr. Pharmacol. Rep. 2021, 7, 135–149. [Google Scholar] [CrossRef]
- Zhou, T.; Zhang, T.; Sun, J.; Zhu, H.; Zhang, M.; Wang, X. Tissue-specific transcriptome for Rheum tanguticum reveals candidate genes related to the anthraquinones biosynthesis. Physiol. Mol. Biol. Plants 2021, 27, 2487–2501. [Google Scholar] [CrossRef]
- Bringmann, G.; Irmer, A. Acetogenic anthraquinones: Biosynthetic convergence and chemical evidence of enzymatic cooperation in nature. Phytochem. Rev. 2008, 7, 499–511. [Google Scholar] [CrossRef]
- Bernatoniene, J.; Kopustinskiene, D.M. The role of catechins in cellular responses to oxidative stress. Molecules 2018, 23, 965. [Google Scholar] [CrossRef]
- Lei, H.; Niu, T.; Song, H.; Bai, B.; Liu, A. Comparative transcriptome profiling reveals differentially expressed genes involved in flavonoid biosynthesis between biennial and triennial Sophora flavescens. Ind. Crop. Prod. 2021, 161, 113217. [Google Scholar] [CrossRef]
- Wang, X.; Hu, H.; Wu, Z.; Fan, H.; Wang, G.; Chai, T.; Wang, H. Tissue-specific transcriptome analyses reveal candidate genes for stilbene, flavonoid and anthraquinone biosynthesis in the medicinal plant Polygonum cuspidatum. BMC Genom. 2021, 22, 353. [Google Scholar] [CrossRef]
- Yuan, L.; Pan, K.; Li, Y.; Yi, B.; Gao, B. Comparative transcriptome analysis of Alpinia oxyphylla Miq. reveals tissue-specific expression of flavonoid biosynthesis genes. BMC Genom. Data 2021, 22, 19. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Sun, M.; Zhang, W.; Jiang, C.; Teng, J.; Sheng, W.; Li, M.; Zhang, A.; Duan, Y.; Xue, J. Comparative transcriptome analysis of roots, stems and leaves of Isodon amethystoides reveals candidate genes involved in Wangzaozins biosynthesis. BMC Plant Biol. 2018, 18, 272. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Sun, C.; Shi, F.; Ma, S.; Zheng, J.; Du, X.; Zhang, L. Comparative transcriptome analysis reveals sesquiterpenoid biosynthesis among 1-, 2- and 3-year old Atractylodes chinensis. BMC Plant Biol. 2021, 21, 354. [Google Scholar] [CrossRef]
- Li, S.F.; Zhang, G.J.; Zhang, X.J.; Yuan, J.H.; Deng, C.L.; Gao, W.J. Comparative transcriptome analysis reveals differentially expressed genes associated with sex expression in garden asparagus (Asparagus officinalis). BMC Plant Biol. 2017, 17, 143. [Google Scholar] [CrossRef] [PubMed]
- Choudhri, P.; Rani, M.; Sangwan, R.S.; Kumar, R.; Kumar, A.; Chhokar, V. De novo sequencing, assembly and characterisation of Aloe vera transcriptome and analysis of expression profiles of genes related to saponin and anthraquinone metabolism. BMC Genom. 2018, 19, 427. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Tang, N.; Xu, F.; Chen, Z.; Zhang, X.; Ye, J.; Liao, Y.; Zhang, W.; Kim, S.U.; Wu, P.; et al. SMRT and Illumina RNA sequencing reveal the complexity of terpenoid biosynthesis in Zanthoxylum armatum. Tree Physiol. 2022, 42, 664–683. [Google Scholar] [CrossRef]
- Liu, J.; Leng, L.; Liu, Y.; Gao, H.; Yang, W.; Chen, S.; Liu, A. Identification and quantification of target metabolites combined with transcriptome of two rheum species focused on anthraquinone and flavonoids biosynthesis. Sci. Rep. 2020, 10, 20241. [Google Scholar] [CrossRef]
- Kang, S.H.; Lee, W.H.; Sim, J.S.; Thaku, N.; Chang, S.; Hong, J.P.; Oh, T.J. De novo transcriptome assembly of Senna occidentalis sheds light on the anthraquinone biosynthesis pathway. Front. Plant Sci. 2021, 12, 773553. [Google Scholar] [CrossRef]
- Wang, W.; Wu, B.; Liu, Z.; Zhou, L.; Sun, X.; Tian, J.; Yang, A. Development of EST-SSRs from the ark shell (Scapharca broughtonii) transcriptome and their application in genetic analysis of four populations. Genes Genom. 2021, 43, 669–677. [Google Scholar] [CrossRef]
- Li, H.; Hei, X.; Li, Y.; Wang, G.; Xu, J.; Cheng, S.; Shen, X.; Gao, j.; Yan, Y.; Zhang, G. HPLC analysis of accumulation characteristics of 10 components in different parts of Rheum officinale with different years. China Tradit. Herb. Drugs 2019, 50, 1690–1697. [Google Scholar] [CrossRef]
- Zou, L.; Zhou, N.; Xie, J.; Li, W.; Xiang, D.; Xie, G. Content analysis of five anthraquinone derivatives in different parts of cultivated Rheum officinale. Guangdong Agric. Sci. 2012, 39, 118–122. [Google Scholar] [CrossRef]
- Zhou, T.; Li, Z.H.; Bai, G.Q.; Feng, L.; Chen, C.; Wei, Y.; Chang, Y.X.; Zhao, G.F. Transcriptome sequencing and development of genic SSR markers of an endangered Chinese endemic genus Dipteronia Oliver (Aceraceae). Molecules 2016, 21, 166. [Google Scholar] [CrossRef] [PubMed]
- Ingvarsson, P.K.; Bernhardsson, C. Genome-wide signatures of environmental adaptation in European aspen (Populus tremula) under current and future climate conditions. Evol. Appl. 2020, 13, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Pitts, N.L.; Mudron, M.R.; Durica, D.S.; Mykles, D.L. Transcriptome analysis of the molting gland (Y-organ) from the blackback land crab, Gecarcinus lateralis. Comp. Biochem. Physiol. Part D Genom. Proteom. 2016, 17, 26–40. [Google Scholar] [CrossRef]
- Prakash, A.; Jeffryes, M.; Bateman, A.; Finn, R.D. The HMMER web server for protein sequence similarity search. Curr. Protoc. Bioinformatics 2017, 60, 3–15. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12, 323. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Jozefczuk, J.; Adjaye, J. Quantitative real-time PCR-based analysis of gene expression. Methods Enzymol. 2011, 500, 99–109. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.E.; Wilson, R.K.; Mardis, E.R. The next-generation sequencing revolution and its impact on genomics. Cell 2013, 155, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Pai, T.W.; Li, K.H.; Yang, C.H.; Hu, C.H.; Lin, H.J.; Wang, W.D.; Chen, Y.R. Multiple model species selection for transcriptomics analysis of non-model organisms. BMC Bioinform. 2018, 19, 284. [Google Scholar] [CrossRef]
- Rama Reddy, N.R.; Mehta, R.H.; Soni, P.H.; Makasana, J.; Gajbhiye, N.A.; Ponnuchamy, M.; Kumar, J. Next generation sequencing and transcriptome analysis predicts biosynthetic pathway of Sennosides from Senna (Cassia angustifolia Vahl.), a non-model plant with potent laxative properties. PLoS ONE 2015, 10, e0129422. [Google Scholar] [CrossRef]
- Ungaro, A.; Pech, N.; Martin, J.F.; McCairns, R.J.S.; Mévy, J.P.; Chappaz, R.; Gilles, A. Challenges and advances for transcriptome assembly in non-model species. PLoS ONE 2017, 12, e0185020. [Google Scholar] [CrossRef]
- Li, H.; Zhang, N.; Li, Y.; He, X. High-throughput transcriptomic sequencing of Rheum palmatum L. seedlings and elucidation of genes in anthraquinone biosynthesis. Acta Pharm. Sin. 2018, 53, 1908–1917. [Google Scholar] [CrossRef]
- Mora-Márquez, F.; Chano, V.; Vázquez-Poletti, J.L.; López de Heredia, U. TOA: A software package for automated functional annotation in non-model plant species. Mol. Ecol. Resour. 2021, 21, 621–636. [Google Scholar] [CrossRef]
- Ponniah, S.K.; Thimmapuram, J.; Bhide, K.; Kalavacharla, V.K.; Manoharan, M. Comparative analysis of the root transcriptomes of cultivated sweetpotato (Ipomoea batatas [L.] Lam) and its wild ancestor (Ipomoea trifida [Kunth] G. Don). BMC Plant Biol. 2017, 17, 9. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Pandey, R.P.; Lee, C.M.; Sim, J.S.; Jeong, J.T.; Choi, B.S.; Jung, M.; Ginzburg, D.; Zhao, K.; Won, S.Y.; et al. Genome-enabled discovery of anthraquinone biosynthesis in Senna tora. Nat. Commun. 2020, 11, 5875. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, S.; Mei, Y.; Cai, S.; Gu, Y.; Sun, M.; Liang, Z.; Xiao, Y.; Zhang, M.; Yang, S. A high-quality genome assembly of Morinda officinalis, a famous native southern herb in the Lingnan region of southern China. Hortic. Res. 2021, 8, 135. [Google Scholar] [CrossRef]
- Yu, D.; Xu, F.; Zeng, J.; Zhan, J. Type III polyketide synthases in natural product biosynthesis. IUBMB Life 2012, 64, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Amnuaykanjanasin, A.; Phonghanpot, S.; Sengpanich, N.; Cheevadhanarak, S.; Tanticharoen, M. Insect-specific polyketide synthases (PKSs), potential PKS-nonribosomal peptide synthetase hybrids, and novel PKS clades in tropical fungi. Appl. Environ. Microb. 2009, 75, 3721–3732. [Google Scholar] [CrossRef] [PubMed]
- Abe, I.; Morita, H. Structure and function of the chalcone synthase superfamily of plant type III polyketide synthases. Nat. Prod. Rep. 2010, 27, 809–838. [Google Scholar] [CrossRef]
- Austin, M.B.; Noel, J.P. The chalcone synthase superfamily of type III polyketide synthases. Nat. Prod. Rep. 2003, 20, 79–110. [Google Scholar] [CrossRef]
- Roy, K.; Chanfreau, G.F. A global function for transcription factors in assisting RNA polymerase II termination. Transcription 2018, 9, 41–46. [Google Scholar] [CrossRef]
- Jha, P.; Ochatt, S.J.; Kumar, V. WUSCHEL: A master regulator in plant growth signaling. Plant Cell Rep. 2020, 39, 431–444. [Google Scholar] [CrossRef]
- Li, X.Y.; Wang, Y.; Dai, Y.; He, Y.; Li, C.X.; Mao, P.; Ma, X.R. The transcription factors of tall fescue in response to temperature stress. Plant Biol. 2021, 23, 89–99. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhang, J.; Liu, X.; Meng, M.; Wang, J.; Lin, J. Tissue-specific transcriptome for Dendrobium officinale reveals genes involved in flavonoid biosynthesis. Genomics 2020, 112, 1781–1794. [Google Scholar] [CrossRef] [PubMed]
- Barco, B.; Clay, N.K. Hierarchical and dynamic regulation of defense-responsive specialized metabolism by WRKY and MYB transcription factors. Front. Plant Sci. 2019, 10, 1775. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.; Yu, Y.; Dong, C.; Yang, Y.; Zhai, Y.; Du, F.; Xia, C.; Ni, Z.; Kong, X.; Zhang, L. The bZIP transcription factor TabZIP15 improves salt stress tolerance in wheat. Plant Biotechnol. J. 2021, 19, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, S.; Chen, P.; Cai, J.; Tang, S.; Yang, W.; Cao, F.; Zheng, P.; Sun, B. Systematic analysis of the R2R3-MYB family in camellia sinensis: Evidence for galloylated catechins biosynthesis regulation. Front. Plant Sci. 2021, 12, 782220. [Google Scholar] [CrossRef]
- Green Etxabe, A.; Pini, J.M.; Short, S.; Cunha, L.; Kille, P.; Watson, G.J. Identifying conserved polychaete molecular markers of metal exposure: Comparative analyses using the Alitta virens (Annelida, Lophotrochozoa) transcriptome. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2021, 240, 108913. [Google Scholar] [CrossRef]
- Xu, Y.; Tian, S.; Li, R.; Huang, X.; Li, F.; Ge, F.; Huang, W.; Zhou, Y. Transcriptome characterization and identification of molecular markers (SNP, SSR, and Indels) in the medicinal plant Sarcandra glabra spp. Biomed. Res. Int. 2021, 2021, 9990910. [Google Scholar] [CrossRef]
- Aggarwal, R.K.; Hendre, P.S.; Varshney, R.K.; Bhat, P.R.; Krishnakumar, V.; Singh, L. Identification, characterization and utilization of EST-derived genic microsatellite markers for genome analyses of coffee and related species. Theor. Appl. Genet. 2007, 114, 359–372. [Google Scholar] [CrossRef]
- Jhanwar, S.; Priya, P.; Garg, R.; Parida, S.K.; Tyagi, A.K.; Jain, M. Transcriptome sequencing of wild chickpea as a rich resource for marker development. Plant Biotechnol. J. 2012, 10, 690–702. [Google Scholar] [CrossRef]
- Gupta, S.; Prasad, M. Development and characterization of genic SSR markers in Medicago truncatula and their transferability in leguminous and non-leguminous species. Genome 2009, 52, 761–771. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Transcripts | Unigenes |
|---|---|---|
| Total reads | 1,296,299 | 443,725 |
| Total number | 463,056 | 236,031 |
| GC content | 44.30 | 45.83 |
| N50 value | 1675 | 769 |
| Min length | 179 | 201 |
| Mean length | 839.83 | 563.98 |
| Max length | 24,213 | 24.213 |
| Sum of lengths | 388,889,822 | 133,117,539 |
| Database | Annotated Unigenes | Percentage (%) | 300 ≤ Length < 1000 | Length ≥ 1000 |
|---|---|---|---|---|
| Nr annotation | 126,539 | 53.61 | 48,920 | 20,538 |
| COG annotation | 41,568 | 17.61 | 15,480 | 8508 |
| KOG annotation | 74,696 | 31.65 | 30,882 | 14,500 |
| GO annotation | 20,133 | 8.53 | 7357 | 1952 |
| KEGG annotation | 36,789 | 15.59 | 14,891 | 7889 |
| Swiss-Prot annotation | 56,876 | 24.10 | 23,563 | 13,690 |
| Pfam annotation | 75,256 | 31.88 | 28,886 | 17,752 |
| TrEMBL annotation | 102,507 | 43.43 | 40,253 | 28,366 |
| All annotations | 136,329 | 57.76 | 52,961 | 21,734 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Sun, J.; Zhang, T.; Tang, Y.; Liu, J.; Gao, C.; Zhai, Y.; Guo, Y.; Feng, L.; Zhang, X.; et al. Comparative Transcriptome Analyses of Different Rheum officinale Tissues Reveal Differentially Expressed Genes Associated with Anthraquinone, Catechin, and Gallic Acid Biosynthesis. Genes 2022, 13, 1592. https://doi.org/10.3390/genes13091592
Zhou L, Sun J, Zhang T, Tang Y, Liu J, Gao C, Zhai Y, Guo Y, Feng L, Zhang X, et al. Comparative Transcriptome Analyses of Different Rheum officinale Tissues Reveal Differentially Expressed Genes Associated with Anthraquinone, Catechin, and Gallic Acid Biosynthesis. Genes. 2022; 13(9):1592. https://doi.org/10.3390/genes13091592
Chicago/Turabian StyleZhou, Lipan, Jiangyan Sun, Tianyi Zhang, Yadi Tang, Jie Liu, Chenxi Gao, Yunyan Zhai, Yanbing Guo, Li Feng, Xinxin Zhang, and et al. 2022. "Comparative Transcriptome Analyses of Different Rheum officinale Tissues Reveal Differentially Expressed Genes Associated with Anthraquinone, Catechin, and Gallic Acid Biosynthesis" Genes 13, no. 9: 1592. https://doi.org/10.3390/genes13091592
APA StyleZhou, L., Sun, J., Zhang, T., Tang, Y., Liu, J., Gao, C., Zhai, Y., Guo, Y., Feng, L., Zhang, X., Zhou, T., & Wang, X. (2022). Comparative Transcriptome Analyses of Different Rheum officinale Tissues Reveal Differentially Expressed Genes Associated with Anthraquinone, Catechin, and Gallic Acid Biosynthesis. Genes, 13(9), 1592. https://doi.org/10.3390/genes13091592