
Causes and Pathophysiology of Acquired Sideroblastic Anemia



Abstract
:1. Introduction
2. Historical Context
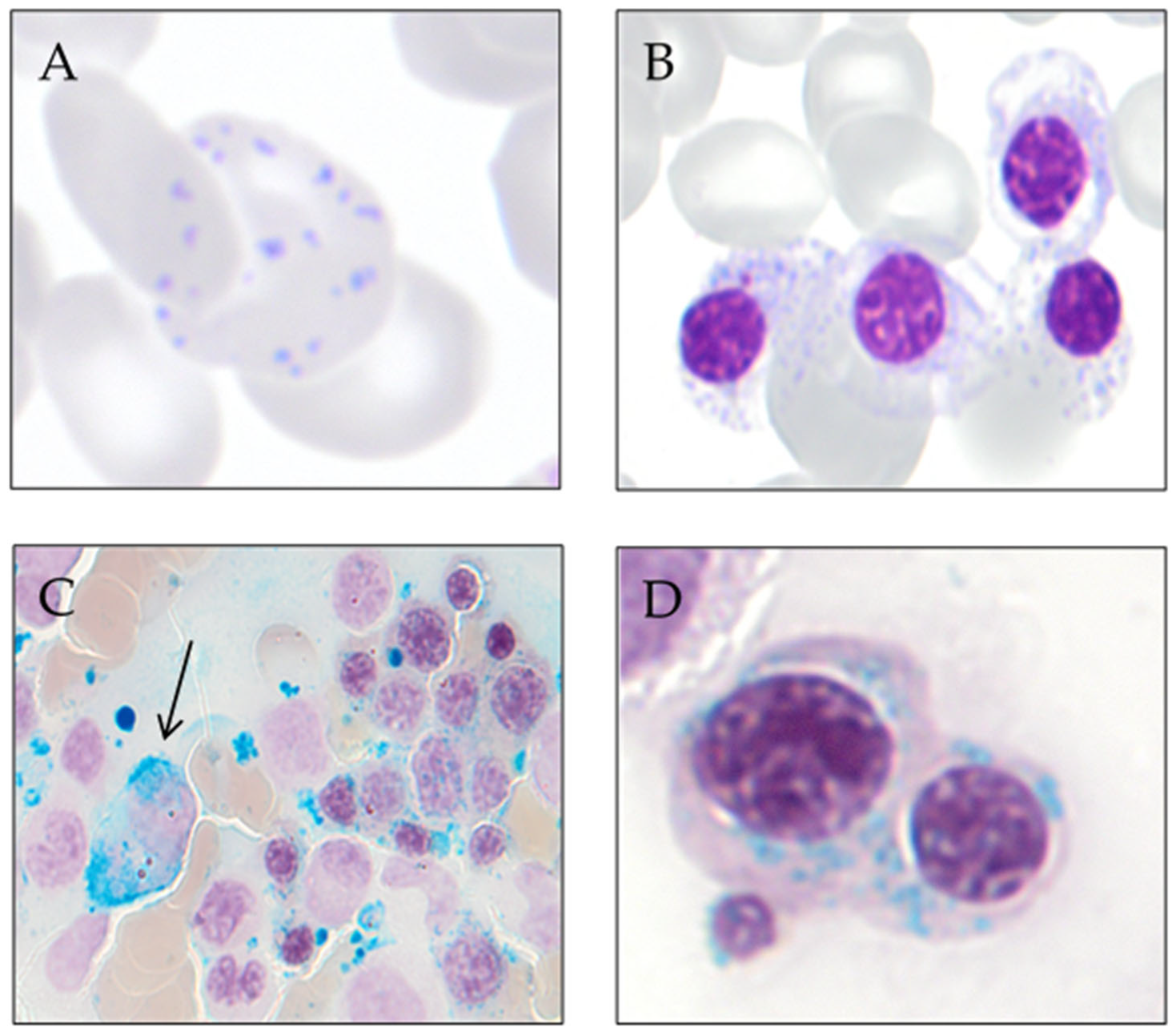
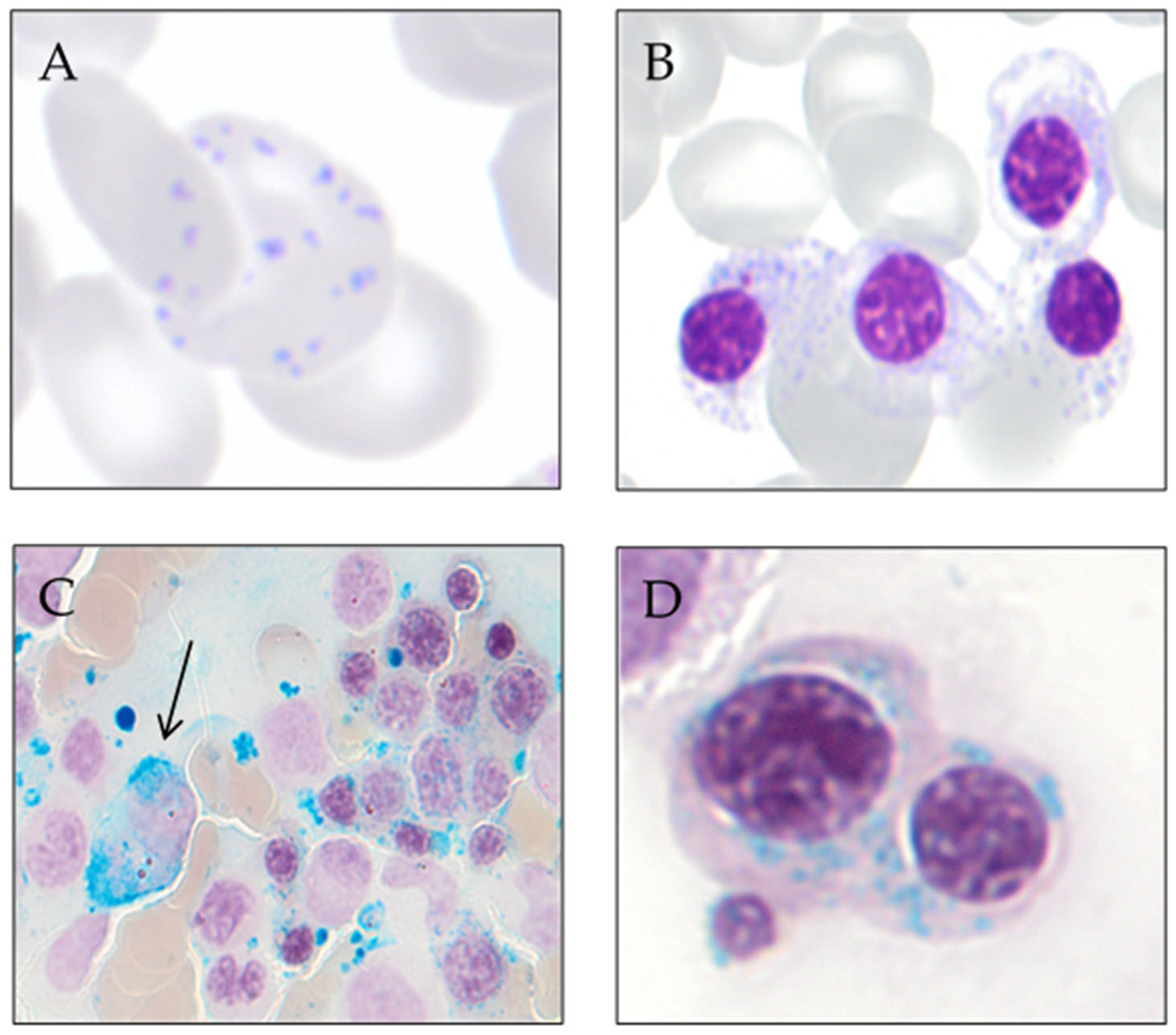
3. Ring Sideroblast Definition
- -
- Type 1 sideroblasts: < 5 siderotic granules in the cytoplasm.
- -
- Type 2 sideroblasts: ≥ 5 siderotic granules, but no perinuclear distribution.
- -
- Type 3 or ring sideroblasts: ≥ 5 siderotic granules in a perinuclear position, covering at least one-third of the nuclear circumference.
4. Classification of Sideroblastic Anemias
5. Ring Sideroblast Formation
5.1. Heme Synthesis
5.2. ISC Biogenesis
5.3. Mitochondrial Respiratory Complex Proteins and Mitochondrial Protein Synthesis
6. Diagnosis of Sideroblastic Anemias
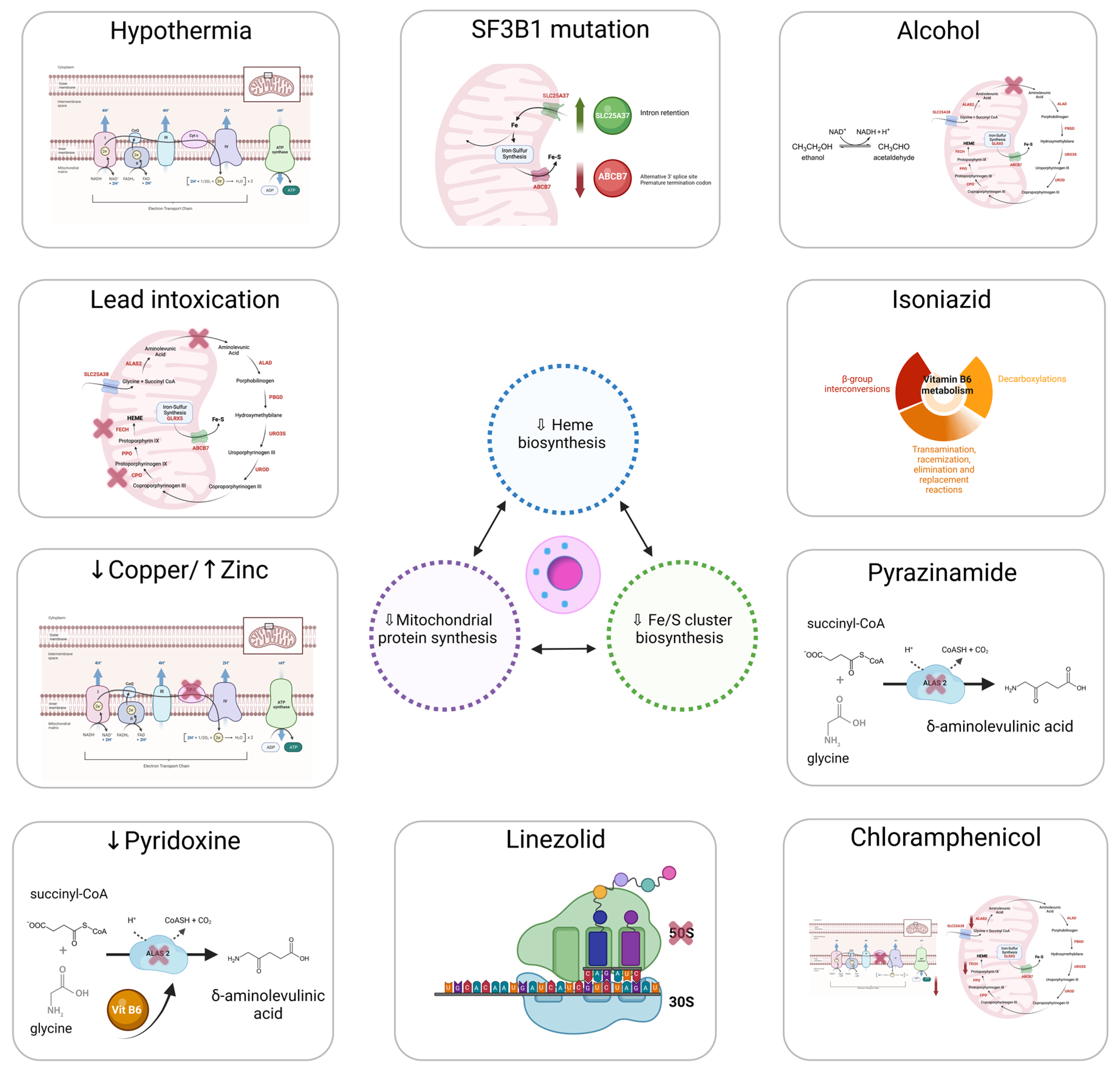
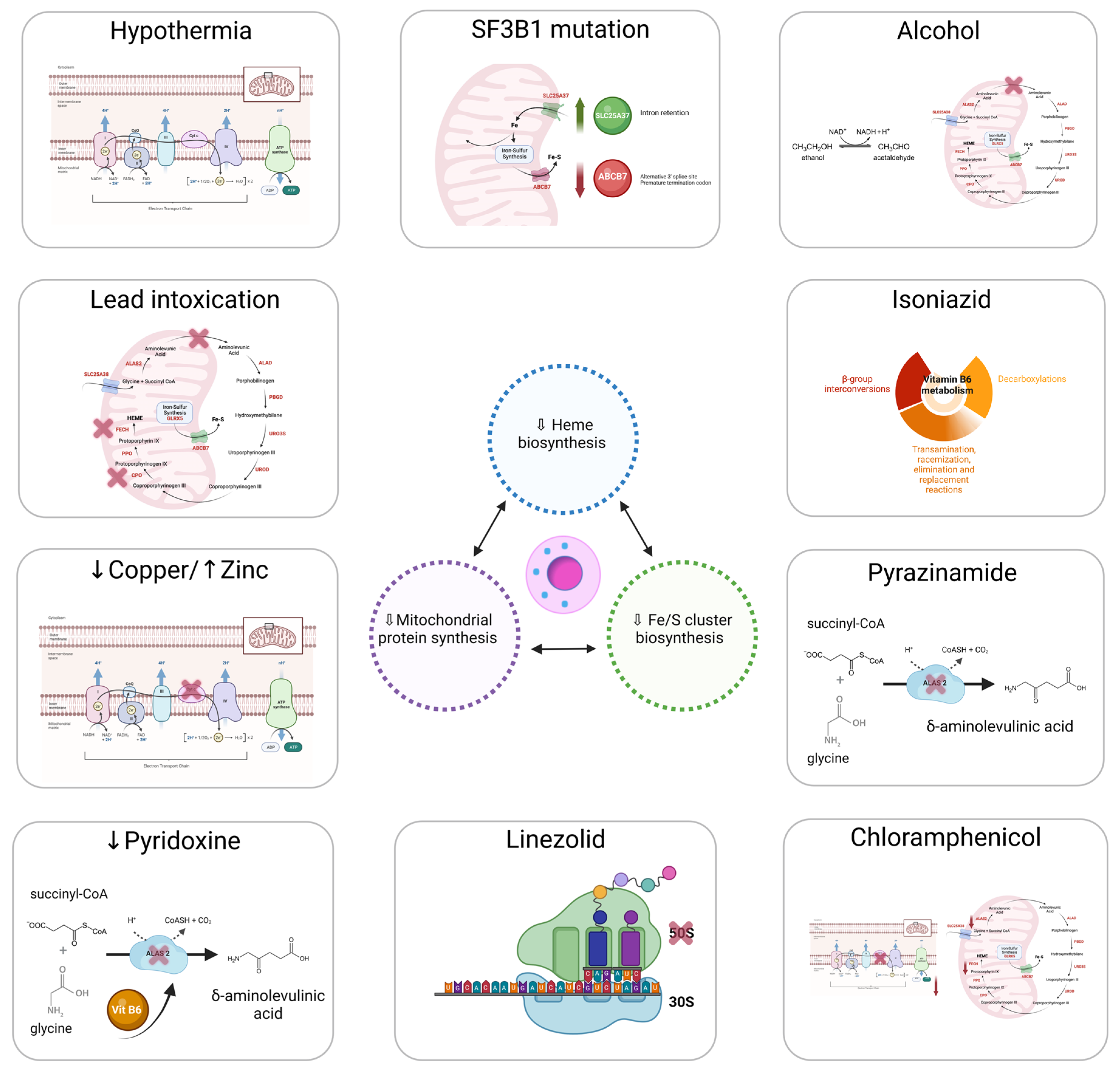
7. Non-Clonal or Metabolic Acquired Sideroblastic Anemias
7.1. Alcohol Consumption
7.2. Drugs
7.2.1. Isoniazid
7.2.2. Pyrazinamide
7.2.3. Chloramphenicol
7.2.4. Linezolid
7.3. Copper Deficiency
7.4. Pyridoxine (Vitamin B6) Deficiency
7.5. Lead Intoxication
7.6. Hypothermia
8. Clonal Sideroblastic Anemias
8.1. Myelodysplastic Syndromes with Ring Sideroblasts (MDS-RS)
8.1.1. Definition
8.1.2. Epidemiology
8.1.3. Clinical Features
8.1.4. Microscopy
8.1.5. Genetic and Molecular Profile
- -
- Cytopenia as defined by standard hematologic values,
- -
- Somatic SF3B1 mutation,
- -
- Morphologic dysplasia (erythroid or multilineage dysplasia), with or without ring sideroblasts,
- -
- Bone marrow blasts <5% and peripheral blood blasts <1%,
- -
- Not meet WHO criteria for MDS with isolated del(5q), MDS/MPN-RS-T or other MDS/MPNs, and primary myelofibrosis or other MPNs,
- -
- Normal karyotype or any cytogenetic abnormality other than del(5q); monosomy 7; and inv(3) or abnormal 3q26, complex (≥3),
- -
- Any additional somatically mutated gene other than RUNX1 and/or EZH2 can be present.
8.1.6. Prognosis
- -
- SF3B15q for concomitant presence with isolated del(5q) (7% of SF3B1-mutant);
- -
- SF3Bb as the commutation between SF3B1 and any gene from BCOR, BCORL1, NRAS, RUNX1, SRSF2, or STAG2 (15% of SF3B1-mutant);
- -
- SF3B1a as any other mutant SF3B1 (78% of SF3B1-mutant), with 107 patients (19%).
8.1.7. Treatment
8.2. Myelodysplastic Syndrome/Myeloproliferative Neoplasm with Ring Sideroblasts and Thrombocytosis (MDS/MPN-RS-T)
8.2.1. Definition
8.2.2. Epidemiology
8.2.3. Clinical Features
8.2.4. Microscopy
8.2.5. Genetic and Molecular Profile
8.2.6. Prognosis
8.2.7. Treatment
8.3. Other Myeloid Neoplasms with Ring Sideroblasts
8.3.1. Primary Myelofibrosis
8.3.2. Acute Myeloid Leukemia
9. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ducamp, S.; Fleming, M.D. The molecular genetics of sideroblastic anemia. Blood 2019, 133, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Grüneberg, H. Siderocytes: A New Kind of Erythrocytes. Nature 1941, 148, 114–115. [Google Scholar] [CrossRef]
- Cooley, T. A severe type of hereditary anemia with elliptocytosis. Interesting sequences of splenectomy. Am. J. Med. Sci. 1945, 209, 561–568. [Google Scholar] [CrossRef]
- Cotter, P.D.; Rucknagel, D.L.; Bishop, D.F. X-linked sideroblastic anemia: Identification of the mutation in the erythroid-specific delta-aminolevulinate synthase gene (ALAS2) in the original family described by Cooley. Blood 1994, 84, 3915–3924. [Google Scholar] [CrossRef]
- Bjorkman, S.E. Chronic refractory anemia with sideroblastic bone marrow; a study of four cases. Blood 1956, 11, 250–259. [Google Scholar] [CrossRef]
- Mollin, D.L. A symposium on sideroblastic anaemia held at the Postgraduate Medical School of London on Friday, March 20th, 1964, during the Annual Meeting of the British Society for Haematology. Introduction: Sideroblasts and Sideroblastic Anaemia. Br. J. Haematol. 1965, 11, 41–48. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; World Health Organization. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2017. [Google Scholar]
- Dharwadkar, A.; Vimal, S.; Panicker, N.K.; Chandanwale, S.S.; Viswanathan, V.; Kumar, H. Study of sideroblasts and iron stores in bone marrow aspirates using Perls′ stain. Med. J. Dr. DY Patil Univ. 2016, 9, 181. [Google Scholar] [CrossRef]
- Juneja, S.K.; Imbert, M.; Jouault, H.; Scoazec, J.Y.; Sigaux, F.; Sultan, C. Haematological features of primary myelodysplastic syndromes (PMDS) at initial presentation: A study of 118 cases. J. Clin. Pathol. 1983, 36, 1129–1135. [Google Scholar] [CrossRef]
- Mufti, G.J.; Bennett, J.M.; Goasguen, J.; Bain, B.J.; Baumann, I.; Brunning, R.; Cazzola, M.; Fenaux, P.; Germing, U.; Hellström-Lindberg, E.; et al. Diagnosis and classification of myelodysplastic syndrome: International Working Group on Morphology of myelodysplastic syndrome (IWGM-MDS) consensus proposals for the definition and enumeration of myeloblasts and ring sideroblasts. Haematologica 2008, 93, 1712–1717. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2008; Volume 2. [Google Scholar]
- Abu-Zeinah, G.; DeSancho, M.T. Understanding Sideroblastic Anemia: An Overview of Genetics, Epidemiology, Pathophysiology and Current Therapeutic Options. J. Blood Med. 2020, 11, 305–318. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Minakami, S.; Yoshikawa, H. Studies on erythrocyte glycolysis. II. Free energy changes and rate limitings steps in erythrocyte glycolysis. J. Biochem. 1966, 59, 139–144. [Google Scholar] [CrossRef]
- Jansen, R.P. Origin and persistence of the mitochondrial genome. Hum. Reprod. 2000, 15, 1–10. [Google Scholar] [CrossRef]
- Zardoya, R. Recent advances in understanding mitochondrial genome diversity. F1000Research 2020, 9, 270. [Google Scholar] [CrossRef]
- McCormick, E.M.; Muraresku, C.C.; Falk, M.J. Mitochondrial Genomics: A Complex Field Now Coming of Age. Curr. Genet. Med. Rep. 2018, 6, 52–61. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Sulaiman, S.A.; Rani, Z.M.; Radin, F.Z.M.; Murad, N.A.A. Advancement in the diagnosis of mitochondrial diseases. J. Transl. Genet. Genom. 2020, 4, 159–187. [Google Scholar] [CrossRef]
- Kuroiwa, T. The discovery of the division apparatus of plastids and mitochondria. QJM Int. J. Med. 2000, 49, 123–134. [Google Scholar] [CrossRef]
- Broker, S.; Meunier, B.; Rich, P.; Gattermann, N.; Hofhaus, G. MtDNA mutations associated with sideroblastic anaemia cause a defect of mitochondrial cytochrome c oxidase. JBIC J. Biol. Inorg. Chem. 1998, 258, 132–138. [Google Scholar] [CrossRef]
- Gattermann, N.; Retzlaff, S.; Wang, Y.L.; Hofhaus, G.; Heinisch, J.; Aul, C.; Schneider, W. Heteroplasmic point mutations of mitochondrial DNA affecting subunit I of cytochrome c oxidase in two patients with acquired idiopathic sideroblastic anemia. Blood 1997, 90, 4961–4972. [Google Scholar] [CrossRef]
- Rascati, R.; Parsons, P. Purification and characterization of cytochrome c oxidase from rat liver mitochondria. J. Biol. Chem. 1979, 254, 1586–1593. [Google Scholar] [CrossRef]
- Chen, J.-J. Regulation of protein synthesis by the heme-regulated eIF2α kinase: Relevance to anemias. Blood 2006, 109, 2693–2699. [Google Scholar] [CrossRef]
- Bottomley, S.S.; Muller-Eberhard, U. Pathophysiology of heme synthesis. Semin. Hematol. 1988, 25, 282–302. [Google Scholar]
- Fujiwara, T.; Harigae, H. Biology of Heme in Mammalian Erythroid Cells and Related Disorders. BioMed Res. Int. 2015, 2015, 278536. [Google Scholar] [CrossRef]
- Rudd, D.M. Elsevier’s Integrated Review Biochemistry, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Ponka, P. Tissue-Specific Regulation of Iron Metabolism and Heme Synthesis: Distinct Control Mechanisms in Erythroid. Cells Blood 1997, 89, 1–25. [Google Scholar] [CrossRef]
- Mustajoki, S.; Laine, M.; Lahtela, M.; Mustajoki, P.; Peltonen, L.; Kauppinen, R. Acute Intermittent Porphyria: Expression of Mutant and Wild-Type Porphobilinogen Deaminase in COS-1 Cells. Mol. Med. 2000, 6, 670–679. [Google Scholar] [CrossRef]
- Rademakers, L.H.P.M.; Koningsberger, J.C.; Sorber, C.W.J.; DE LA Faille, H.B.; VAN Hattum, J.; Marx, J.J.M. Accumulation of iron in erythroblasts of patients with erythropoietic protoporphyria. Eur. J. Clin. Investig. 1993, 23, 130–138. [Google Scholar] [CrossRef]
- Lecha, M.; Puy, H.; Deybach, J.C. Erythropoietic protoporphyria. Orphanet. J. Rare Dis. 2009, 4, 19. [Google Scholar] [CrossRef]
- Bishop, D.F.; Henderson, A.S.; Astrin, K.H. Human delta-aminolevulinate synthase: Assignment of the housekeeping gene to 3p21 and the erythroid-specific gene to the X chromosome. Genomics 1990, 7, 207–214. [Google Scholar] [CrossRef]
- Riddle, R.D.; Yamamoto, M.; Engel, J.D. Expression of delta-aminolevulinate synthase in avian cells: Separate genes encode erythroid-specific and nonspecific isozymes. Proc. Natl. Acad. Sci. USA 1989, 86, 792–796. [Google Scholar] [CrossRef]
- Cotter, P.D.; Willard, H.F.; Gorski, J.L.; Bishop, D.F. Assignment of human erythroid delta-aminolevulinate synthase (ALAS2) to a distal subregion of band Xp11.21 by PCR analysis of somatic cell hybrids containing X; autosome translocations. Genomics 1992, 13, 211–212. [Google Scholar] [CrossRef]
- Cox, T.C.; Bawden, M.J.; Martin, A.; May, B.K. Human erythroid 5-aminolevulinate synthase: Promoter analysis and identification of an iron-responsive element in the mRNA. EMBO J. 1991, 10, 1891–1902. [Google Scholar] [CrossRef]
- Bergmann, A.K.; Bs, D.R.C.; BS, E.M.M.; Agarwal, S.; Fleming, M.D.; Bottomley, S.S.; Neufeld, E.J. Systematic molecular genetic analysis of congenital sideroblastic anemia: Evidence for genetic heterogeneity and identification of novel mutations. Pediatr. Blood Cancer 2009, 54, 273–278. [Google Scholar] [CrossRef]
- Stehling, O.; Lill, R. The Role of Mitochondria in Cellular Iron-Sulfur Protein Biogenesis: Mechanisms, Connected Processes, and Diseases. Cold Spring Harb. Perspect. Biol. 2013, 5, a011312. [Google Scholar] [CrossRef]
- Bottomley, S.S.; Fleming, M.D. Sideroblastic anemias: Molecular basis, pathophysiology, and clinical aspects. In Handbook of Porphyrin Science with Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine; Porphyrias and Sideroblastic Anemias, World Scientific: Singapore, 2014; Volume 29, pp. 43–87. [Google Scholar]
- Zhang, D.-L.; Ghosh, M.C.; Rouault, T.A. The physiological functions of iron regulatory proteins in iron homeostasis—An update. Front. Pharmacol. 2014, 5, 124. [Google Scholar] [CrossRef]
- Hamdi, A.; Roshan, T.M.; Kahawita, T.M.; Mason, A.B.; Sheftel, A.D.; Ponka, P. Erythroid cell mitochondria receive endosomal iron by a “kiss-and-run” mechanism. Biochim. Biophys. Acta 2016, 1863, 2859–2867. [Google Scholar] [CrossRef]
- Camaschella, C.; Nai, A.; Silvestri, L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020, 105, 260–272. [Google Scholar] [CrossRef]
- Drysdale, J.; Arosio, P.; Invernizzi, R.; Cazzola, M.; Volz, A.; Corsi, B.; Biasiotto, G.; Levi, S. Mitochondrial Ferritin: A New Player in Iron Metabolism. Blood Cells Mol. Dis. 2002, 29, 376–383. [Google Scholar] [CrossRef]
- Bou-Abdallah, F.; Santambrogio, P.; Levi, S.; Arosio, P.; Chasteen, N.D. Unique Iron Binding and Oxidation Properties of Human Mitochondrial Ferritin: A Comparative Analysis with Human H-chain Ferritin. J. Mol. Biol. 2005, 347, 543–554. [Google Scholar] [CrossRef]
- Nie, G.; Sheftel, A.D.; Kim, S.F.; Ponka, P.; Brown, P.; Levis, M.; Shurtleff, S.; Campana, D.; Downing, J.; Small, D. Overexpression of mitochondrial ferritin causes cytosolic iron depletion and changes cellular iron homeostasis. Blood 2005, 105, 2161–2167. [Google Scholar] [CrossRef]
- Bessis, M.C.; Breton-Gorius, J. Ferritin and Ferruginous Micelles in Normal Erythroblasts and Hypochromic Hypersideremic Anemias. Blood 1959, 14, 423–432. [Google Scholar] [CrossRef]
- Levi, S.; Corsi, B.; Bosisio, M.; Invernizzi, R.; Volz, A.; Sanford, D.; Arosio, P.; Drysdale, J. A Human Mitochondrial Ferritin Encoded by an Intronless Gene. J. Biol. Chem. 2001, 276, 24437–24440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazzola, M.; Invernizzi, R.; Bergamaschi, G.; Levi, S.; Corsi, B.; Travaglino, E.; Rolandi, V.; Biasiotto, G.; Drysdale, J.; Arosio, P. Mitochondrial ferritin expression in erythroid cells from patients with sideroblastic anemia. Blood 2003, 101, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Liochev, S.I.; Fridovich, I. The role of O2.- in the production of HO.: In vitro and in vivo. Free Radic. Biol. Med. 1994, 16, 29–33. [Google Scholar] [CrossRef]
- Gutteridge, J.M.; Rowley, D.A.; Halliwell, B. Superoxide-dependent formation of hydroxyl radicals in the presence of iron salts. Detection of ‘free’ iron in biological systems by using bleomycin-dependent degradation of DNA. Biochem. J. 1981, 199, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Barrientos, T.; Andrews, N.C. Iron and Copper in Mitochondrial Diseases. Cell Metab. 2013, 17, 319–328. [Google Scholar] [CrossRef]
- Bottomley, S.S.; Fleming, M.D. Sideroblastic anemia: Diagnosis and management. Hematol. Oncol. Clin. N. Am. 2014, 28, 653–670. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C. Hereditary Sideroblastic Anemias: Pathophysiology, Diagnosis, and Treatment. Semin. Hematol. 2009, 46, 371–377. [Google Scholar] [CrossRef]
- Tanno, T.; Miller, J.L. Iron Loading and Overloading due to Ineffective Erythropoiesis. Adv. Hematol. 2010, 2010, 358283. [Google Scholar] [CrossRef]
- Ohba, R.; Furuyama, K.; Yoshida, K.; Fujiwara, T.; Fukuhara, N.; Onishi, Y.; Manabe, A.; Ito, E.; Ozawa, K.; Kojima, S.; et al. Clinical and genetic characteristics of congenital sideroblastic anemia: Comparison with myelodysplastic syndrome with ring sideroblast (MDS-RS). Ann. Hematol. 2012, 92, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zahid, M.F.; Khan, N.; Pei, J.; Testa, J.R.; Dulaimi, E. Genomic imbalances in peripheral blood confirm the diagnosis of myelodysplastic syndrome in a patient presenting with non-immune hemolytic anemia. Leuk. Res. Rep. 2016, 5, 23–26. [Google Scholar] [CrossRef]
- Woessner, S.F.L. La Citología Óptica en el Diagnóstico Hematológico, 5th ed.; Acción Médica: Madrid, Spain, 2006. [Google Scholar]
- Acín, P.; Florensa, L.; Andreu, L.; Woessner, S. Cytoplasmic abnormalities of erythroblasts as a marker for ringed sideroblasts in myelodysplastic syndromes. Eur. J. Haematol. 2009, 54, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.A.; Lobel, J.S.; Kocoshis, S.A.; Naiman, J.L.; Windmiller, J.; Lammi, A.T.; Hoffman, R.; Marsh, J.C. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J. Pediatr. 1979, 95, 976–984. [Google Scholar] [CrossRef]
- Ballard, H.S. The hematological complications of alcoholism. Alcohol Health Res. World 1997, 21, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Sieg, I.; Doss, M.O.; Kandels, H.; Schneider, J. Effect of alcohol on delta-aminolevulinic acid dehydratase and porphyrin metabolism in man. Clin. Chim. Acta Int. J. Clin. Chem. 1991, 202, 211–218. [Google Scholar] [CrossRef]
- Eichner, E.R.; Hillman, R.S. The evolution of anemia in alcoholic patients. Am. J. Med. 1971, 50, 218–232. [Google Scholar] [CrossRef]
- Savage, D.; Lindenbaum, J. Anemia in alcoholics. Medicine 1986, 65, 322–338. [Google Scholar] [CrossRef]
- Pierce, H.I.; McGuffin, R.G.; Hillman, R.S. Clinical studies in alcoholic sideroblastosis. Arch. Intern. Med. 1976, 136, 283–289. [Google Scholar] [CrossRef]
- Tenner, S.; Rollhauser, C.; Butt, F.; Gonzalez, P. Sideroblastic anemia. A diagnosis to consider in alcoholic patients. Postgrad. Med. 1992, 92, 147–150. [Google Scholar] [CrossRef]
- Mangla, G.; Garg, N.; Bansal, D.; Kotru, M.; Sikka, M. Peripheral Blood and Bone Marrow Findings in Chronic Alcoholics with Special Reference to Acquired Sideroblastic Anemia. Indian J. Hematol. Blood Transfus. 2020, 36, 559–564. [Google Scholar] [CrossRef]
- Latvala, J.; Parkkila, S.; Melkko, J.; Niemelä, O. Acetaldehyde Adducts in Blood and Bone Marrow of Patients With Ethanol-Induced Erythrocyte Abnormalities. Mol. Med. 2001, 7, 401–405. [Google Scholar] [CrossRef]
- Azhar, W.; Zaidi, F.; Hannan, A. Isoniazid Induced Pure Red Blood Cell Aplasia. Cureus 2020, 12, e7112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fratz-Berilla, E.J.; Breydo, L.; Gouya, L.; Puy, H.; Uversky, V.N.; Ferreira, G.C. Isoniazid inhibits human erythroid 5-aminolevulinate synthase: Molecular mechanism and tolerance study with four X-linked protoporphyria patients. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1863, 428–439. [Google Scholar] [CrossRef]
- Verwilghen, R.; Reybrouck, G.; Callens, L.; Cosemans, J. Antituberculous Drugs and Sideroblastic Anaemia. Br. J. Haematol. 1965, 11, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Murali, N.; Permi, V.D.; Shetty, A.K. Sideroblastic Anemia Associated with Isoniazid Prophylaxis in a Person Living with HIV. Am. J. Ther. 2019, 27, e409–e410. [Google Scholar] [CrossRef]
- Minardi, M.; Fato, I.; Di Gennaro, F.; Mosti, S.; Mastrobattista, A.; Cerva, C.; Libertone, R.; Saracino, A.; Goletti, D.; Girardi, E.; et al. Common and Rare Hematological Manifestations and Adverse Drug Events during Treatment of Active TB: A State of Art. Microorganisms 2021, 9, 1477. [Google Scholar] [CrossRef]
- Colucci, G.; Silzle, T.; Solenthaler, M. Pyrazinamide-induced sideroblastic anemia. Am. J. Hematol. 2011, 87, 22125. [Google Scholar] [CrossRef] [PubMed]
- Leiter, L.M.; Thatte, H.S.; Okafor, C.; Marks, P.W.; Golan, D.E.; Bridges, K.R. Chloramphenicol-induced mitochondrial dysfunction is associated with decreased transferrin receptor expression and ferritin synthesis in K562 cells and is unrelated to IRE-IRP interactions. J. Cell. Physiol. 1999, 180, 334–344. [Google Scholar]
- Beck, E.; Ziegler, G.; Schmid, R.; Lüdin, H. Reversible Sideroblastic Anemia Caused by Chloramphenicol. Acta Haematol. 1967, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.; Marcus, O. Effect of chloramphenicol on reticulocyte delta-aminolaevulinic acid synthetase in rabbits. Br. J. Haematol. 1974, 26, 79–83. [Google Scholar] [CrossRef]
- Ammus, S.; Yunis, A. Drug-induced red cell dyscrasias. Blood Rev. 1989, 3, 71–82. [Google Scholar] [CrossRef]
- Soriano, A.; Miró, O.; Mensa, J. Mitochondrial Toxicity Associated with Linezolid. N. Engl. J. Med. 2005, 353, 2305–2306. [Google Scholar] [CrossRef] [PubMed]
- Leach, K.L.; Swaney, S.M.; Colca, J.R.; McDonald, W.G.; Blinn, J.R.; Thomasco, L.M.; Gadwood, R.C.; Shinabarger, D.; Xiong, L.; Mankin, A.S. The Site of Action of Oxazolidinone Antibiotics in Living Bacteria and in Human Mitochondria. Mol. Cell 2007, 26, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, W.B.; Trotta, R.F.; Rector, J.T.; A Tjaden, J.; Barile, A.J. Mechanisms for Linezolid-Induced Anemia and Thrombocytopenia. Ann. Pharmacother. 2003, 37, 517–520. [Google Scholar] [CrossRef]
- Willekens, C.; Dumézy, F.; Boyer, T.; Renneville, A.; Rossignol, J.; Berthon, C.; Cotteau-Leroy, A.; Mehiaoui, L.; Quesnel, B.; Preudhomme, C. Linezolid induces ring sideroblasts. Haematologica 2013, 98, e138–e140. [Google Scholar] [CrossRef]
- Liapis, K.; Vrachiolias, G.; Spanoudakis, E.; Kotsianidis, I. Vacuolation of early erythroblasts with ring sideroblasts: A clue to the diagnosis of linezolid toxicity. Br. J. Haematol. 2020, 190, 809. [Google Scholar] [CrossRef] [PubMed]
- Cousins, R.J. Absorption, transport, and hepatic metabolism of copper and zinc: Special reference to metallothionein and ceruloplasmin. Physiol. Rev. 1985, 65, 238–309. [Google Scholar] [CrossRef] [PubMed]
- Wahab, A.; Mushtaq, K.; Borak, S.G.; Bellam, N. Zinc-induced copper deficiency, sideroblastic anemia, and neutropenia: A perplexing facet of zinc excess. Clin. Case Rep. 2020, 8, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Monaghan, S.A.; Miller, M.L.; McKenna, R.W.; Perkins, W.D.; Levinson, B.S.; Bhushan, V.; Kroft, S.H. Zinc-induced copper deficiency: A report of three cases initially recognized on bone marrow examination. Am. J. Clin. Pathol. 2005, 123, 125–131. [Google Scholar]
- Gregg, X.T.; Reddy, V.; Prchal, J.T. Copper deficiency masquerading as myelodysplastic syndrome. Blood 2002, 100, 1493–1495. [Google Scholar] [CrossRef]
- D’Angelo, G. Copper deficiency mimicking myelodysplastic syndrome. Blood Res. 2016, 51, 217–219. [Google Scholar] [CrossRef]
- Halfdanarson, T.R.; Kumar, N.; Li, C.-Y.; Phyliky, R.L.; Hogan, W.J. Hematological manifestations of copper deficiency: A retrospective review. Eur. J. Haematol. 2008, 80, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Fong, T.; Vij, R.; Vijayan, A.; DiPersio, J.; Blinder, M. Copper deficiency: An important consideration in the differential diagnosis of myelodysplastic syndrome. Haematologica 2007, 92, 1429–1430. [Google Scholar] [CrossRef] [PubMed]
- Karri, S.; Doshi, V. Hematological Abnormalities in Copper Deficiency. Blood 2007, 110, 2677. [Google Scholar] [CrossRef]
- Kumar, N.; Crum, B.; Petersen, R.C.; Vernino, S.; Ahlskog, J.E. Copper Deficiency Myelopathy. Arch. Neurol. 2004, 61, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Lazarchick, J. Update on anemia and neutropenia in copper deficiency. Curr. Opin. Hematol. 2012, 19, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Villalba, A.; Senent, L. Differential diagnosis of myelodysplastic syndrome: Anemia associated with copper deficiency. Blood 2018, 131, 1389. [Google Scholar] [CrossRef]
- Pirruccello, E.; Luu, H.S.; Chen, W. Haematogone hyperplasia in copper deficiency. Br. J. Haematol. 2016, 173, 335. [Google Scholar] [CrossRef]
- Sutton, L.; Vusirikala, M.; Chen, W. Hematogone Hyperplasia in Copper Deficiency. Am. J. Clin. Pathol. 2009, 132, 191–199. [Google Scholar] [CrossRef]
- Whittaker, M.M.; Penmatsa, A.; Whittaker, J.W. The Mtm1p carrier and pyridoxal 5′-phosphate cofactor trafficking in yeast mitochondria. Arch. Biochem. Biophys. 2015, 568, 64–70. [Google Scholar] [CrossRef]
- Vogler, W.R.; Mingioli, E.S. Heme Synthesis in Pyridoxine-Responsive Anemia. N. Engl. J. Med. 1965, 273, 347–353. [Google Scholar] [CrossRef]
- Narang, N.C.; Kotru, M.; Rao, K.; Sikka, M. Megaloblastic Anemia with Ring Sideroblasts is not Always Myelodysplastic Syndrome. Turk. J. Haematol. 2016, 33, 358–359. [Google Scholar] [CrossRef] [PubMed]
- Knollmann-Ritschel, B.E.C.; Markowitz, M. Educational Case: Lead Poisoning. Acad. Pathol. 2017, 4, e4916. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.; Royal, J.; Seeler, R. Interaction of iron deficiency and lead and the hematologic findings in children with severe lead poisoning. Pediatrics 1988, 81, 247–254. [Google Scholar]
- Bhambhani, K.; Aronow, R. Lead poisoning and thalassemia trait or iron deficiency. The value of the red blood cell distribution width. Am. J. Dis. Child. 1990, 144, 1231–1233. [Google Scholar] [PubMed]
- Lubran, M.M. Lead toxicity and heme biosynthesis. Ann. Clin. Lab. Sci. 1980, 10, 402–413. [Google Scholar]
- Connell, N.T.; Benz, E.J. Sideroblastic anemias. In Anemia: Pathophysiology, Diagnosis, and Management; Benz, J.E.J., Schiffman, F.J., Berliner, N., Eds.; Cambridge University Press: Cambridge, UK, 2017; pp. 44–47. [Google Scholar]
- O’Brien, H.; Amess, J.A.L.; Mollin, D.L. Recurrent thrombocytopenia, erythroid hypoplasia and sideroblastic anaemia associated with hypothermia. Br. J. Haematol. 1982, 51, 451–456. [Google Scholar] [CrossRef]
- Eicher, D.J.; Wagner, C.L.; Katikaneni, L.P.; Hulsey, T.C.; Bass, W.T.; Kaufman, D.A.; Horgan, M.J.; Languani, S.; Bhatia, J.J.; Givelichian, L.M.; et al. Moderate hypothermia in neonatal encephalopathy: Safety outcomes. Pediatr. Neurol. 2005, 32, 18–24. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Wyatt, J.S.; Azzopardi, D.; Ballard, R.; Edwards, A.D.; Ferriero, D.M.; Polin, R.A.; Robertson, C.M.; Thoresen, M.; Whitelaw, A.; et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: Multicentre randomised trial. Lancet 2005, 365, 663–670. [Google Scholar]
- Lemyre, B.; Chau, V. Hypothermia for newborns with hypoxic-ischemic encephalopathy. Paediatr. Child Health 2018, 23, 285–291. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemia: Integrating Morphological, Clinical, and Genomic Data. Blood 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Germing, U.; Gattermann, N.; Aivado, M.; Hildebrandt, B.; Aul, C. Two types of acquired idiopathic sideroblastic anaemia (AISA): A time-tested distinction. Br. J. Haematol. 2000, 108, 724–728. [Google Scholar] [CrossRef]
- Malcovati, L.; Della Porta, M.G.; Pascutto, C.; Invernizzi, R.; Boni, M.; Travaglino, E.; Passamonti, F.; Arcaini, L.; Maffioli, M.; Bernasconi, P.; et al. Prognostic Factors and Life Expectancy in Myelodysplastic Syndromes Classified According to WHO Criteria: A Basis for Clinical Decision Making. J. Clin. Oncol. 2005, 23, 7594–7603. [Google Scholar] [CrossRef]
- Malcovati, L.; Germing, U.; Kuendgen, A.; Della Porta, M.G.; Pascutto, C.; Invernizzi, R.; Giagounidis, A.; Hildebrandt, B.; Bernasconi, P.; Knipp, S.; et al. Time-Dependent Prognostic Scoring System for Predicting Survival and Leukemic Evolution in Myelodysplastic Syndromes. J. Clin. Oncol. 2007, 25, 3503–3510. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Lasho, T.L.; Hodnefield, J.M.; Knudson, R.A.; Ketterling, R.P.; Garcia-Manero, G.; Steensma, D.P.; Pardanani, A.; Hanson, C.A.; Tefferi, A. SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood 2012, 119, 569–572. [Google Scholar] [CrossRef]
- Germing, U.; Gattermann, N.; Strupp, C.; Aivado, M.; Aul, C. Validation of the WHO proposals for a new classification of primary myelodysplastic syndromes: A retrospective analysis of 1600 patients. Leuk. Res. 2000, 24, 983–992. [Google Scholar] [CrossRef]
- Ambaglio, I.; Malcovati, L.; Papaemmanuil, E.; Laarakkers, C.M.; Della Porta, M.G.; Gallì, A.; Da Vià, M.C.; Bono, E.; Ubezio, M.; Travaglino, E.; et al. Inappropriately low hepcidin levels in patients with myelodysplastic syndrome carrying a somatic mutation of SF3B1. Haematologica 2013, 98, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Ambaglio, I.; Pinto, V.; Girelli, D.; Elena, C.; Campostrini, N.; Pietra, D.; Galli, A.; Travaglino, E.; Cazzola, M.; Forni, G.L.; et al. SF3B1 Mutation Is an Independent Predictor of Parenchymal Iron Overload in Myelodysplastic Syndromes. Blood 2015, 126, 1678. [Google Scholar] [CrossRef]
- Germing, U.; Strupp, C.; Kuendgen, A.; Isa, S.; Knipp, S.; Hildebrandt, B.; Giagounidis, A.; Aul, C.; Gattermann, N.; Haas, R. Prospective validation of the WHO proposals for the classification of myelodysplastic syndromes. Haematologica 2006, 91, 1596–1604. [Google Scholar]
- Malcovati, L.; Karimi, M.; Papaemmanuil, E.; Ambaglio, I.; Jädersten, M.; Jansson, M.; Elena, C.; Gallì, A.; Walldin, G.; Della Porta, M.G.; et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood 2015, 126, 233–241. [Google Scholar] [CrossRef]
- Margolskee, E.; Hasserjian, R.P.; Hassane, D.; Tam, W.; Mathew, S.; Ok, C.Y.; Wang, S.A.; Oak, J.; Arber, D.A.; Orazi, A. Myelodysplastic Syndrome, Unclassifiable (MDS-U) with 1% Blasts Is a Distinct Subgroup of MDS-U with a Poor Prognosis. Am. J. Clin. Pathol. 2017, 148, 49–57. [Google Scholar] [CrossRef]
- Schanz, J.; Tüchler, H.; Solé, F.; Mallo, M.; Luño, E.; Cervera, J.; Granada, I.; Hildebrandt, B.; Slovak, M.L.; Ohyashiki, K.; et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J. Clin. Oncol. 2012, 30, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.A.; Abruzzo, L.V.; Hasserjian, R.P.; Zhang, L.; Hu, Y.; Zhang, Y.; Zhao, M.; Galili, N.; Raza, A.; Medeiros, L.J.; et al. Myelodysplastic syndromes with deletions of chromosome 11q lack cryptic MLL rearrangement and exhibit characteristic clinicopathologic features. Leuk. Res. 2011, 35, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. SomaticSF3B1Mutation in Myelodysplasia with Ring Sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Adema, V.; Khouri, J.; Ni, Y.; Rogers, H.J.; Kerr, C.M.; Awada, H.; Nagata, Y.; Kuzmanovic, T.; Advani, A.S.; Gerds, A.T.; et al. Analysis of distinct SF3B1 hotspot mutations in relation to clinical phenotypes and response to therapy in myeloid neoplasia. Leuk. Lymphoma 2020, 62, 735–738. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Malcovati, L.; Gallì, A.; Sato-Otsubo, A.; Kataoka, K.; Sato, Y.; Watatani, Y.; Suzuki, H.; Yoshizato, T.; Yoshida, K.; et al. Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat. Commun. 2018, 9, 3649. [Google Scholar] [CrossRef] [Green Version]
- Dolatshad, H.; Pellagatti, A.; Fernandez-Mercado, M.; Yip, B.H.; Malcovati, L.; Attwood, M.; Przychodzen, B.; Sahgal, N.; Kanapin, A.A.; Lockstone, H.; et al. Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells. Leukemia 2015, 29, 1092–1103. [Google Scholar] [CrossRef]
- Conte, S.; Katayama, S.; Vesterlund, L.; Karimi, M.; Dimitriou, M.; Jansson, M.; Mortera-Blanco, T.; Unneberg, P.; Papaemmanuil, E.; Sander, B.; et al. Aberrant splicing of genes involved in haemoglobin synthesis and impaired terminal erythroid maturation in SF3B1 mutated refractory anaemia with ring sideroblasts. Br. J. Haematol. 2015, 171, 478–490. [Google Scholar] [CrossRef]
- Visconte, V.; Rogers, H.J.; Singh, J.; Barnard, J.; Bupathi, M.; Traina, F.; McMahon, J.; Makishima, H.; Szpurka, H.; Jankowska, A.; et al. SF3B1 haploinsufficiency leads to formation of ring sideroblasts in myelodysplastic syndromes. Blood 2012, 120, 3173–3186. [Google Scholar] [CrossRef]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G.; et al. Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 2016, 30, 404–417. [Google Scholar] [CrossRef] [PubMed]
- Boultwood, J.; Pellagatti, A.; Nikpour, M.; Pushkaran, B.; Fidler, C.; Cattan, H.; Littlewood, T.J.; Malcovati, L.; Della Porta, M.G.; Jädersten, M.; et al. The role of the iron transporter ABCB7 in refractory anemia with ring sideroblasts. PLoS ONE 2008, 3, e1970. [Google Scholar] [CrossRef]
- Pellagatti, A.; Cazzola, M.; Giagounidis, A.A.; Malcovati, L.; Porta, M.G.; Killick, S.; Campbell, L.J.; Wang, L.; Langford, C.F.; Fidler, C.; et al. Gene expression profiles of CD34+ cells in myelodysplastic syndromes: Involvement of interferon-stimulated genes and correlation to FAB subtype and karyotype. Blood 2006, 108, 337–345. [Google Scholar] [CrossRef]
- Harigae, H.; Nakajima, O.; Suwabe, N.; Yokoyama, H.; Furuyama, K.; Sasaki, T.; Kaku, M.; Yamamoto, M.; Sassa, S. Aberrant iron accumulation and oxidized status of erythroid-specific δ-aminolevulinate synthase (ALAS2)–deficient definitive erythroblasts. Blood 2003, 101, 1188–1193. [Google Scholar] [CrossRef]
- Pondarre, C.; Campagna, D.R.; Antiochos, B.; Sikorski, L.; Mulhern, H.; Fleming, M. Abcb7, the gene responsible for X-linked sideroblastic anemia with ataxia, is essential for hematopoiesis. Blood 2006, 109, 3567–3569. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.; Reilly, A.; Hayes, B.J.; Clough, C.A.; Konnick, E.Q.; Torok-Storb, B.; Gulsuner, S.; Wu, D.; Becker, P.S.; Keel, S.B.; et al. Reprogramming identifies functionally distinct stages of clonal evolution in myelodysplastic syndromes. Blood 2019, 134, 186–198. [Google Scholar] [CrossRef]
- Mupo, A.; Seiler, M.; Sathiaseelan, V.; Pance, A.; Yang, Y.; A Agrawal, A.; Iorio, F.; Bautista, R.; Pacharne, S.; Tzelepis, K.; et al. Hemopoietic-specific Sf3b1-K700E knock-in mice display the splicing defect seen in human MDS but develop anemia without ring sideroblasts. Leukemia 2016, 31, 720–727. [Google Scholar] [CrossRef]
- Matsunawa, M.; Yamamoto, R.; Sanada, M.; Sato-Otsubo, A.; Shiozawa, Y.; Yoshida, K.; Otsu, M.; Shiraishi, Y.; Miyano, S.; Isono, K.; et al. Haploinsufficiency of Sf3b1 leads to compromised stem cell function but not to myelodysplasia. Leukemia 2014, 28, 1844–1850. [Google Scholar] [CrossRef]
- Malcovati, L.; Stevenson, K.; Papaemmanuil, E.; Neuberg, D.; Bejar, R.; Boultwood, J.; Bowen, D.T.; Campbell, P.J.; Ebert, B.L.; Fenaux, P.; et al. SF3B1-mutant MDS as a distinct disease subtype: A proposal from the International Working Group for the Prognosis of MDS. Blood 2020, 136, 157–170. [Google Scholar] [CrossRef]
- Cazzola, M.; Della Porta, M.G.; Malcovati, L. The genetic basis of myelodysplasia and its clinical relevance. Blood 2013, 122, 4021–4034. [Google Scholar] [CrossRef]
- Visconte, V.; Makishima, H.; Jankowska, A.; Szpurka, H.; Traina, F.; Jerez, A.; O’Keefe, C.; Rogers, H.J.; Sekeres, M.A.; Maciejewski, J.P.; et al. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia 2012, 26, 542–545. [Google Scholar] [CrossRef]
- Malcovati, L.; Papaemmanuil, E.; Bowen, D.T.; Boultwood, J.; Della Porta, M.G.; Pascutto, C.; Travaglino, E.; Groves, M.J.; Godfrey, A.L.; Ambaglio, I.; et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood 2011, 118, 6226–6239. [Google Scholar] [CrossRef] [PubMed]
- Platzbecker, U.; Germing, U.; Götze, K.S.; Kiewe, P.; Mayer, K.; Chromik, J.; Radsak, M.; Wolff, T.; Zhang, X.; Laadem, A.; et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): A multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017, 18, 1338–1347. [Google Scholar] [CrossRef]
- Fenaux, P.; Kiladjian, J.J.; Platzbecker, U. Luspatercept for the treatment of anemia in myelodysplastic syndromes and primary myelofibrosis. Blood 2019, 133, 790–794. [Google Scholar] [CrossRef]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Díez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhan, O.; et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 382, 140–151. [Google Scholar]
- Kurtovic-Kozaric, A.; Przychodzen, B.; Singh, J.A.; Konarska, M.M.; Clemente, M.J.; Otrock, Z.K.; Nakashima, M.; Hsi, E.D.; Yoshida, K.; Shiraishi, Y.; et al. PRPF8 defects cause missplicing in myeloid malignancies. Leukemia 2015, 29, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Swoboda, D.M.; Kanagal-Shamanna, R.; Brunner, A.M.; Cluzeau, T.; Chan, O.; Al Ali, N.; Montalban-Bravo, G.; Gesiotto, Q.J.; Gavralidis, A.; Hunter, A.M.; et al. Marrow ring sideroblasts are highly predictive for TP53 mutation in MDS with excess blasts. Leukemia 2022, 36, 1189–1192. [Google Scholar] [CrossRef]
- Greenberg, P.; Cox, C.; Lebeau, M.M.; Fenaux, P.; Morel, P.; Sanz, G.; Sanz, M.; Vallespi, T.; Hamblin, T.; Oscier, D.; et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997, 89, 2079–2088. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465. [Google Scholar]
- Kantarjian, H.; O’Brien, S.; Ravandi, F.; Cortes, J.; Shan, J.; Bennett, J.M.; List, A.; Fenaux, P.; Sanz, G.; Issa, J.-P.; et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer 2008, 113, 1351–1361. [Google Scholar] [CrossRef]
- Breccia, M.; Carmosino, I.; Biondo, F.; Mancini, M.; Russo, E.; Latagliata, R.; Alimena, G. Usefulness and prognostic impact on survival of WHO reclassification in FAB low risk myelodyplastic syndromes. Leuk. Res. 2006, 30, 178–182. [Google Scholar] [CrossRef]
- Li, Y.; Cui, R.; Qin, T.; Xu, Z.; Zhang, Y.; Cai, W.; Cui, W.; Liu, J.; Li, B.; Xiao, Z. Validation of the WHO 2016 proposals for Myelodysplastic syndromes patients with the presence of ring sideroblasts but without excess blasts. Br. J. Haematol. 2016, 178, 813–816. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Hanson, C.A.; Sulai, N.H.; Hodnefield, J.M.; Knudson, R.A.; Ketterling, R.P.; Lasho, T.L.; Tefferi, A. Prognostic irrelevance of ring sideroblast percentage in World Health Organization–defined myelodysplastic syndromes without excess blasts. Blood 2012, 119, 5674–5677. [Google Scholar] [CrossRef]
- Damm, F.; Kosmider, O.; Gelsi-Boyer, V.; Renneville, A.; Carbuccia, N.; Hidalgo-Curtis, C.; Della Valle, V.; Couronné, L.; Scourzic, L.; Chesnais, V.; et al. Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood 2012, 119, 3211–3218. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.E.; Caughey, B.A.; Abdel-Wahab, O.; Steensma, D.P.; Galili, N.; Raza, A.; Kantarjian, H.; Levine, R.L.; Neuberg, D.; et al. Validation of a Prognostic Model and the Impact of Mutations in Patients With Lower-Risk Myelodysplastic Syndromes. J. Clin. Oncol. 2012, 30, 3376–3382. [Google Scholar] [CrossRef]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Ossa, J.E.A.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid. 2022, 1, EVIDoa2200008. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Tefferi, A. Myelodysplastic syndromes with ring sideroblasts (MDS-RS) and MDS/myeloproliferative neoplasm with RS and thrombocytosis (MDS/MPN-RS-T)—“2021 update on diagnosis, risk-stratification, and management”. Am. J. Hematol. 2021, 96, 379–394. [Google Scholar] [CrossRef]
- Hellström-Lindberg, E. Efficacy of erythropoietin in the myelodysplastic syndromes: A meta-analysis of 205 patients from 17 studies. Br. J. Haematol. 1995, 89, 67–71. [Google Scholar] [CrossRef]
- Moyo, V.; Lefebvre, P.; Duh, M.S.; Yektashenas, B.; Mundle, S. Erythropoiesis-stimulating agents in the treatment of anemia in myelodysplastic syndromes: A meta-analysis. Ann. Hematol. 2008, 87, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Reeves, J.A.; Feldman, E.J.; Dewald, G.W.; Bennett, J.M.; Deeg, H.J.; Dreisbach, L.; Schiffer, C.A.; Stone, R.M.; Greenberg, P.L.; et al. Phase 2 study of lenalidomide in transfusion-dependent, low-risk, and intermediate-1 risk myelodysplastic syndromes with karyotypes other than deletion 5q. Blood 2008, 111, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Santini, V.; Almeida, A.; Giagounidis, A.; Gröpper, S.; Jonasova, A.; Vey, N.; Mufti, G.J.; Buckstein, R.; Mittelman, M.; Platzbecker, U.; et al. Randomized Phase III Study of Lenalidomide versus Placebo in RBC Transfusion-Dependent Patients with Lower-Risk Non-del(5q) Myelodysplastic Syndromes and Ineligible for or Refractory to Erythropoiesis-Stimulating Agents. J. Clin. Oncol. 2016, 34, 2988–2996. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Vallet, S. Sotatercept, a soluble activin receptor type 2A IgG-Fc fusion protein for the treatment of anemia and bone loss. Curr. Opin. Mol. Ther. 2010, 12, 586–597. [Google Scholar] [PubMed]
- Cermák, J.; Jonasova, A.; Vondrakova, J.; Walterová, L.; Hochová, I.; Siskova, M.; Neuwirtová, R. Efficacy and Safety Of Administration of Oral Iron Chelator Deferiprone in Patients with Early Myelodysplastic Syndrome. Hemoglobin 2011, 35, 217–227. [Google Scholar] [CrossRef]
- Angelucci, E.; Li, J.; Greenberg, P.; Wu, D.; Hou, M.; Montano Figueroa, E.H.; Rodriguez, M.G.; Dong, X.; Ghosh, J.; Izquierdo, M.; et al. Iron Chelation in Transfusion-Dependent Patients with Low- to Intermediate-1-Risk Myelodysplastic Syndromes: A Randomized Trial. Ann. Intern. Med. 2020, 172, 513–522. [Google Scholar] [CrossRef]
- Garcia-Manero, G. Myelodysplastic syndromes: 2014 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2014, 89, 97–108. [Google Scholar] [CrossRef]
- Morita, Y.; Maeda, Y.; Yamaguchi, T.; Urase, F.; Kawata, S.; Hanamoto, H.; Tsubaki, K.; Ishikawa, J.; Shibayama, H.; Matsumura, I.; et al. Five-day regimen of azacitidine for lower-risk myelodysplastic syndromes (refractory anemia or refractory anemia with ringed sideroblasts): A prospective single-arm phase 2 trial. Cancer Sci. 2018, 109, 3209–3215. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Gore, S.D.; Cogle, C.; Ward, R.; Shi, T.; Macbeth, K.J.; Laille, E.; Giordano, H.; Sakoian, S.; Jabbour, E.; et al. Phase I study of oral azacitidine in myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia. J. Clin. Oncol. 2011, 29, 2521–2527. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, E.; Short, N.J.; Montalban-Bravo, G.; Huang, X.; Bueso-Ramos, C.; Qiao, W.; Yang, H.; Zhao, C.; Kadia, T.; Borthakur, G.; et al. Randomized phase 2 study of low-dose decitabine vs low-dose azacitidine in lower-risk MDS and MDS/MPN. Blood 2017, 130, 1514–1522. [Google Scholar] [CrossRef]
- Savona, M.R.; Odenike, O.; Amrein, P.C.; Steensma, D.P.; E DeZern, A.; Michaelis, L.C.; Faderl, S.; Harb, W.; Kantarjian, H.; Lowder, J.; et al. An oral fixed-dose combination of decitabine and cedazuridine in myelodysplastic syndromes: A multicentre, open-label, dose-escalation, phase 1 study. Lancet Haematol. 2019, 6, e194–e203. [Google Scholar] [CrossRef]
- Steensma, D.P.; Fenaux, P.; Van Eygen, K.; Raza, A.; Santini, V.; Germing, U.; Font, P.; Diez-Campelo, M.; Thepot, S.; Vellenga, E.; et al. Imetelstat Achieves Meaningful and Durable Transfusion Independence in High Transfusion–Burden Patients with Lower-Risk Myelodysplastic Syndromes in a Phase II Study. J. Clin. Oncol. 2021, 39, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Deeg, H.J.; Scott, B.L.; Fang, M.; Shulman, H.M.; Gyurkocza, B.; Myerson, D.; Pagel, J.M.; Platzbecker, U.; Ramakrishnan, A.; Radich, J.P.; et al. Five-group cytogenetic risk classification, monosomal karyotype, and outcome after hematopoietic cell transplantation for MDS or acute leukemia evolving from MDS. Blood 2012, 120, 1398–1408. [Google Scholar] [CrossRef]
- Carré, M.; Porcher, R.; Finke, J.; Ehninger, G.; Koster, L.; Beelen, D.; Ganser, A.; Volin, L.; Lozano, S.; Friis, L.; et al. Role of Age and Hematopoietic Cell Transplantation-Specific Comorbidity Index in Myelodysplastic Patients Undergoing an Allotransplant: A Retrospective Study from the Chronic Malignancies Working Party of the European Group for Blood and Marrow Transplantation. Biol. Blood Marrow Transplant. 2019, 26, 451–457. [Google Scholar] [CrossRef]
- Jaffe, E.S. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues; World Health Organization Classification of Tumours: Geneva, Switzerland, 2001. [Google Scholar]
- Raya, J.M.; Arenillas, L.; Domingo, A.; Bellosillo, B.; Gutiérrez, G.; Luno, E.; Pinan, M.A.; Barbón, M.; Perez-Sirvent, M.L.; Muruzabal, M.J.; et al. Refractory anemia with ringed sideroblasts associated with thrombocytosis: Comparative analysis of marked with non-marked thrombocytosis, and relationship with JAK2 V617F mutational status. Int. J. Hematol. 2008, 88, 387–395. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Lasho, T.L.; Finke, C.M.; Hanson, C.A.; King, R.L.; Ketterling, R.; Gangat, N.; Tefferi, A. Predictors of survival in refractory anemia with ring sideroblasts and thrombocytosis (RARS-T) and the role of next-generation sequencing. Am. J. Hematol. 2016, 91, 492–498. [Google Scholar] [CrossRef]
- Sabine, J.; Torsten, H.; Sandra, W.; Manja, M.; Christiane, E.; Niroshan, N.; Tamara, A.; Alexander, K.; Wolfgang, K.; Claudia, H.; et al. Refractory anemia with ring sideroblasts and marked thrombocytosis cases harbor mutations in SF3B1 or other spliceosome genes accompanied by JAK2V617F and ASXL1 mutations. Haematologica 2015, 100, e125–e127. [Google Scholar]
- Broseus, J.; Florensa, L.; Zipperer, E.; Schnittger, S.; Malcovati, L.; Richebourg, S.; Lippert, E.; Cermak, J.; Evans, J.; Mounier, M.; et al. Clinical features and course of refractory anemia with ring sideroblasts associated with marked thrombocytosis. Haematologica 2012, 97, 1036–1041. [Google Scholar] [CrossRef]
- Broséus, J.; Lippert, E.; Harutyunyan, A.S.; Jeromin, S.; Zipperer, E.; Florensa, L.; Milosevic, J.D.; Haferlach, T.; Germing, U.; Luño, E.; et al. Low rate of calreticulin mutations in refractory anaemia with ring sideroblasts and marked thrombocytosis. Leukemia 2014, 28, 1374–1376. [Google Scholar] [CrossRef] [Green Version]
- Malcovati, L.; Della Porta, M.G.; Pietra, D.; Boveri, E.; Pellagatti, A.; Gallì, A.; Travaglino, E.; Brisci, A.; Rumi, E.; Passamonti, F.; et al. Molecular and clinical features of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Blood 2009, 114, 3538–3545. [Google Scholar] [CrossRef]
- Mangaonkar, A.A.; Lasho, T.L.; Ketterling, R.P.; Reichard, K.K.; Gangat, N.; Al-Kali, A.; Begna, K.H.; Pardanani, A.; Al Ali, N.H.; Talati, C.; et al. Myelodysplastic/myeloproliferative neoplasms with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T): Mayo-Moffitt collaborative study of 158 patients. Blood Cancer J. 2022, 12, 26. [Google Scholar] [CrossRef]
- Antelo, G.; Mangaonkar, A.A.; Coltro, G.; Buradkar, A.; Lasho, T.L.; Finke, C.; Carr, R.; Binder, M.; Gangat, N.; Al-Kali, A.; et al. Response to erythropoiesis-stimulating agents in patients with WHO-defined myelodysplastic syndrome/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T). Br. J. Haematol. 2020, 189, e104–e108. [Google Scholar] [CrossRef] [PubMed]
- Nicolosi, M.; Mudireddy, M.; Vallapureddy, R.; Gangat, N.; Tefferi, A.; Patnaik, M.M. Lenalidomide therapy in patients with myelodysplastic syndrome/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T). Am. J. Hematol. 2017, 93, E27–E30. [Google Scholar] [CrossRef] [PubMed]
- Huls, G.; Mulder, A.B.; Rosati, S.; van de Loosdrecht, A.A.; Vellenga, E.; de Wolf, J.T.M. Efficacy of single-agent lenalidomide in patients with JAK2 (V617F) mutated refractory anemia with ring sideroblasts and thrombocytosis. Blood 2010, 116, 180–182. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Lasho, T.L.; Finke, C.M.; Hanson, C.A.; King, R.L.; Ketterling, R.P.; Gangat, N.; Tefferi, A. Vascular events and risk factors for thrombosis in refractory anemia with ring sideroblasts and thrombocytosis. Leukemia 2016, 30, 2273–2275. [Google Scholar] [CrossRef]
- Lasho, T.L.; Finke, C.M.; Hanson, C.A.; Jimma, T.; Knudson, R.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. SF3B1 mutations in primary myelofibrosis: Clinical, histopathology and genetic correlates among 155 patients. Leukemia 2012, 26, 1135–1137. [Google Scholar] [CrossRef]
- Boiocchi, L.; Hasserjian, R.P.; Pozdnyakova, O.; Wong, W.J.; Lennerz, J.K.; Le, L.P.; Dias-Santagata, D.; Iafrate, A.J.; Hobbs, G.S.; Nardi, V. Clinicopathological and molecular features of SF3B1-mutated myeloproliferative neoplasms. Hum. Pathol. 2018, 86, 1–11. [Google Scholar] [CrossRef]
- Talwar, A.; Bansal, S.; Bapna, A.; Sharma, U. Myelodysplastic syndrome/myeloproliferative neoplasm-ring sideroblast with myelofibrosis—A diagnostic dilemma? A distinct entity. Indian J. Pathol. Microbiol. 2021, 64, 434–436. [Google Scholar]
- Xu, Z. MDS/MPN with ring sideroblasts and thrombocytosis masquerading as prefibrotic/early primary myelofibrosis. Blood 2017, 129, 657. [Google Scholar] [CrossRef]
- Martin-Cabrera, P.; Jeromin, S.; Perglerovà, K.; Haferlach, C.; Kern, W.; Haferlach, T. Acute myeloid leukemias with ring sideroblasts show a unique molecular signature straddling secondary acute myeloid leukemia and de novo acute myeloid leukemia. Haematologica 2017, 102, e125–e128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Berger, G.; Gerritsen, M.; Yi, G.; Koorenhof-Scheele, T.N.; Kroeze, L.; Stevens-Kroef, M.; Yoshida, K.; Shiraishi, Y.; Berg, E.V.D.; Schepers, H.; et al. Ring sideroblasts in AML are associated with adverse risk characteristics and have a distinct gene expression pattern. Blood Adv. 2019, 3, 3111–3122. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Inheritance | Gene | Syndromic | Age at Presentation | Anemia Severity | MCV | Other Symptoms | |
|---|---|---|---|---|---|---|---|
| Congenital | |||||||
| Heme synthesis defects | |||||||
| XLSA | X | ALAS2 (* 301300) | No | Infancy to adulthood | Mild to severe | ↓
 N/↑  | Iron overload in the absence of transfusions |
| SLC25A38 | AR | SLC25A38 (* 610819) | No | Infancy | Severe | ↓ | Transfusional iron overload |
| Erythropoietic protoporphyria | AR/PSD | FECH (* 612386) | No | Childhood | Mild | ↓ | Acute photosensitivity |
| Fe-S biogenesis defects | |||||||
| GLRX5 deficiency | AR | GLRX5 (* 609588) | No | Adulthood | Mild to severe | ↓ | Iron overload |
| HSPA9 deficiency | AR/PSD | HSPA9 (* 600548) | No | Childhood | Mild to severe | N/↓ | Retinitis pigmentosa |
| HSCB deficiency | AR | HSCB (* 608142) | No | Childhood | Moderate | N | None |
| XLSA/A | X | ABCB7 (* 300135) | Yes | Childhood | Mild to moderate | ↓ | Cerebellar ataxia and hypoplasia, delayed motor development |
| Mitochondrial protein synthesis defects | |||||||
| PMPS | SP/M | mtDNA | Yes | Infancy | Severe | ↑ | Lactic acidosis, exocrine pancreatic insufficiency, failure to thrive, hepatic/renal failure |
| MLASA1 | AR | PUS1 (* 608109) | Yes | Childhood | Mild to severe | N/↑ | Myopathy, lactic acidosis, facial dysmorphism |
| MLASA2 | AR | YARS2 (* 610957) | Yes | Childhood | Mild to severe | N/↑ | Myopathy, lactic acidosis, cardiomyopathy |
| LARS2 deficiency | AR | LARS2 (* 604544) | Yes | Infancy | Severe | ↑ | Lactic acidosis, cardiomyopathy, hepatopathy, seizures |
| SIFD | AR | TRNT1 (* 612907) | Yes | Infancy | Severe | ↓ | Immunodeficiency, aseptic febrile episodes, developmental delay, seizures, cardiomyopathy, retinitis pigmentosa, other |
| Mitochondrial protein synthesis defects | |||||||
| MT-ATP6-SA | SP/M | MT-ATP6 (* 516060) | Yes | Infancy to early childhood | Mild to severe | N/↑ | Lactic acidosis, myopathy, neurological abnormalities |
| NDUFB11-SA multifactorial | X | NDUFB11 (* 300403) | Yes | Early childhood | Moderate | N | Lactic acidosis, myopathy |
| TRMA | AR | SLC19A2 (* 603941) | Yes | Early childhood | Mild to severe | ↑ | Sensorineural deafness, non-type-I diabetes mellitus, optic atrophy, stroke-like episodes |
| Acquired | |||||||
| MDS-RS-SLD | Somatic | SF3B1 (* 605590) | N/A | Adulthood | Mild to moderate | ↑/N | Iron overload |
| MDS-RS-MLD | Somatic | SF3B1 | N/A | Adulthood | Mild to moderate | ↑/N | Iron overload, other cytopenias |
| MDS/MPN-RS-T | Somatic | SF3B1 JAK2 (* 147796), CALR (* 109091) o MPL (* 159530) | N/A | Adulthood | Mild | ↑/N | Thrombocytosis |
| Cytopenia/s | Dysplastic Lineages | Blasts | % RS | Others | |
|---|---|---|---|---|---|
| MDS-RS-SLD | 1 or 2 | 1 | <1% PB and <5% BM | ≥15 or ≥5 if SF3B1 mut | No MDS 5q criteria No Auer rods |
| MDS-RS-MLD | 1–3 | ≥2 | <1% PB and 5% BM | ≥15 or ≥5 if SF3B1 mut | No MDS 5q criteria No Auer rods |
| MDS/MPN-RS-T | Anemia | 1–3 | <1% PB and 5% BM | ≥15 | Thrombocytosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez-Sevilla, J.J.; Calvo, X.; Arenillas, L. Causes and Pathophysiology of Acquired Sideroblastic Anemia. Genes 2022, 13, 1562. https://doi.org/10.3390/genes13091562
Rodriguez-Sevilla JJ, Calvo X, Arenillas L. Causes and Pathophysiology of Acquired Sideroblastic Anemia. Genes. 2022; 13(9):1562. https://doi.org/10.3390/genes13091562
Chicago/Turabian StyleRodriguez-Sevilla, Juan Jose, Xavier Calvo, and Leonor Arenillas. 2022. "Causes and Pathophysiology of Acquired Sideroblastic Anemia" Genes 13, no. 9: 1562. https://doi.org/10.3390/genes13091562
APA StyleRodriguez-Sevilla, J. J., Calvo, X., & Arenillas, L. (2022). Causes and Pathophysiology of Acquired Sideroblastic Anemia. Genes, 13(9), 1562. https://doi.org/10.3390/genes13091562