

mintRULS: Prediction of miRNA–mRNA Target Site Interactions Using Regularized Least Square Method

_Guda.png)
Abstract
:
1. Introduction
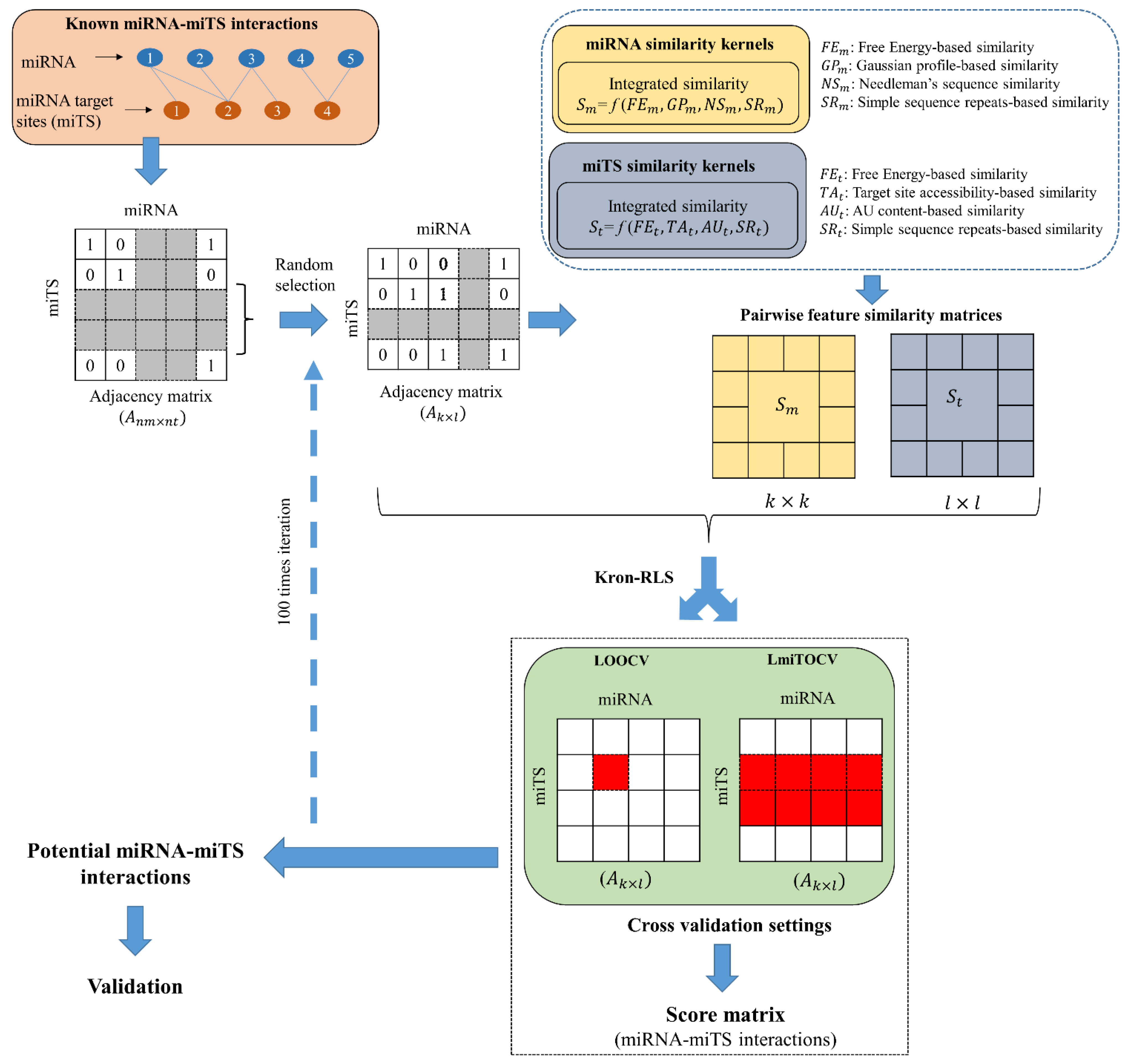
2. Materials and Methods
2.1. miRNA–Target Site Associations in Human and Mouse
2.2. Kernel Similarity Scores for miRNA
2.2.1. Free Energy (FE)-Based Similarity
2.2.2. Gaussian Interaction Profile (GP) Kernel Similarity (Based on Known Associations)
2.2.3. Needleman’s Sequence Similarity
2.2.4. Simple Sequence Repeats (SSRs)-Based Similarity
2.2.5. Integration of miRNA Similarity Scores
2.3. Kernel Similarity Scores for miTS
2.3.1. FE-Based Similarity between miTS
2.3.2. Target Site Accessibility (TA)-Based Similarity
2.3.3. AU Content (AU)-Based Similarity
2.3.4. Simple Sequence Repeats (SSRs)-Based Similarity
2.3.5. Integration of miTS’s Pairwise Similarities
2.4. mintRULS
2.5. Cross-Validations and Performance Testing
2.5.1. Cross-Validations
2.5.2. Score Normalization and Performance Evaluation
- Weak Targets: <lower quartile (25th quartile).
- Moderate Targets: between lower quartile (25th quartile) and upper quartile (75th quartile).
- Strong Targets: >upper quartile (75th quartile).
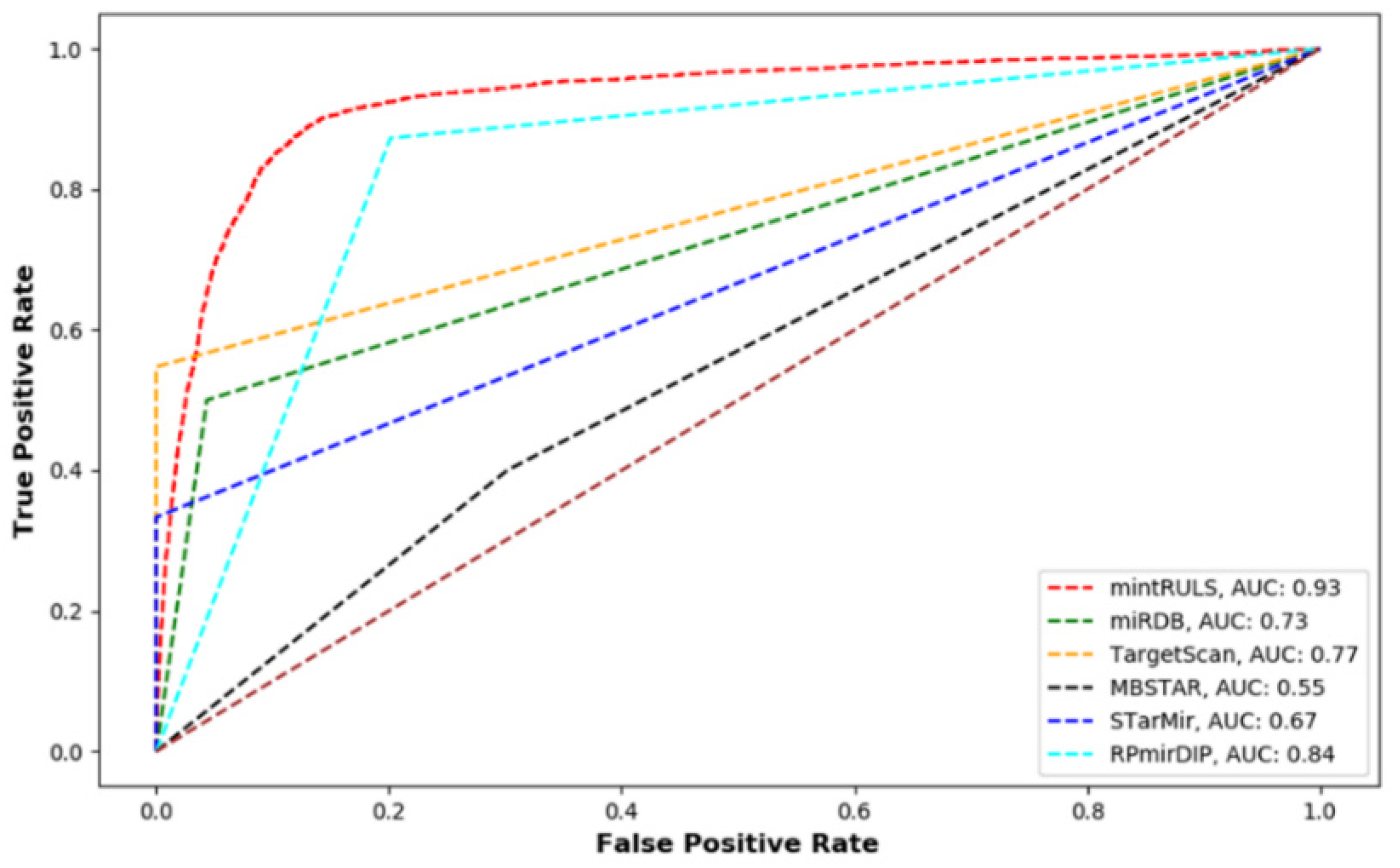
2.5.3. Comparison with Previous Methods
2.6. Model Code Implementation and Software Availability
2.7. Validation of Predictions
2.7.1. Using Condition- and Cell-Specific Studies
2.7.2. Using Literature-Based Data
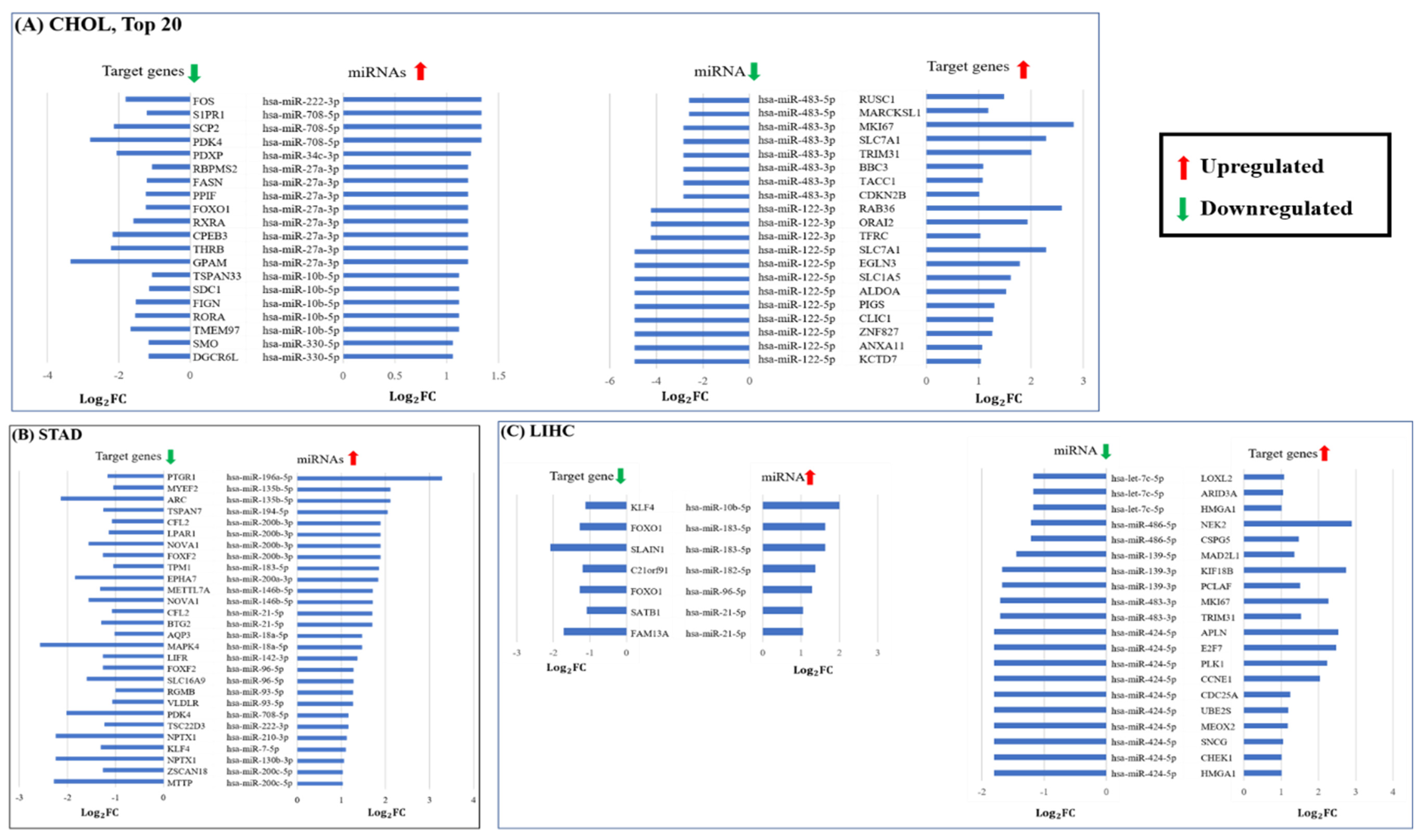
2.7.3. Using Expression Data of miRNA and mRNA in Gastrointestinal (GI) Cancer
2.7.4. Using Expression Data of miRNA and mRNA in Normal and Septic Mice
3. Results
3.1. Performance Evaluation of mintRULS
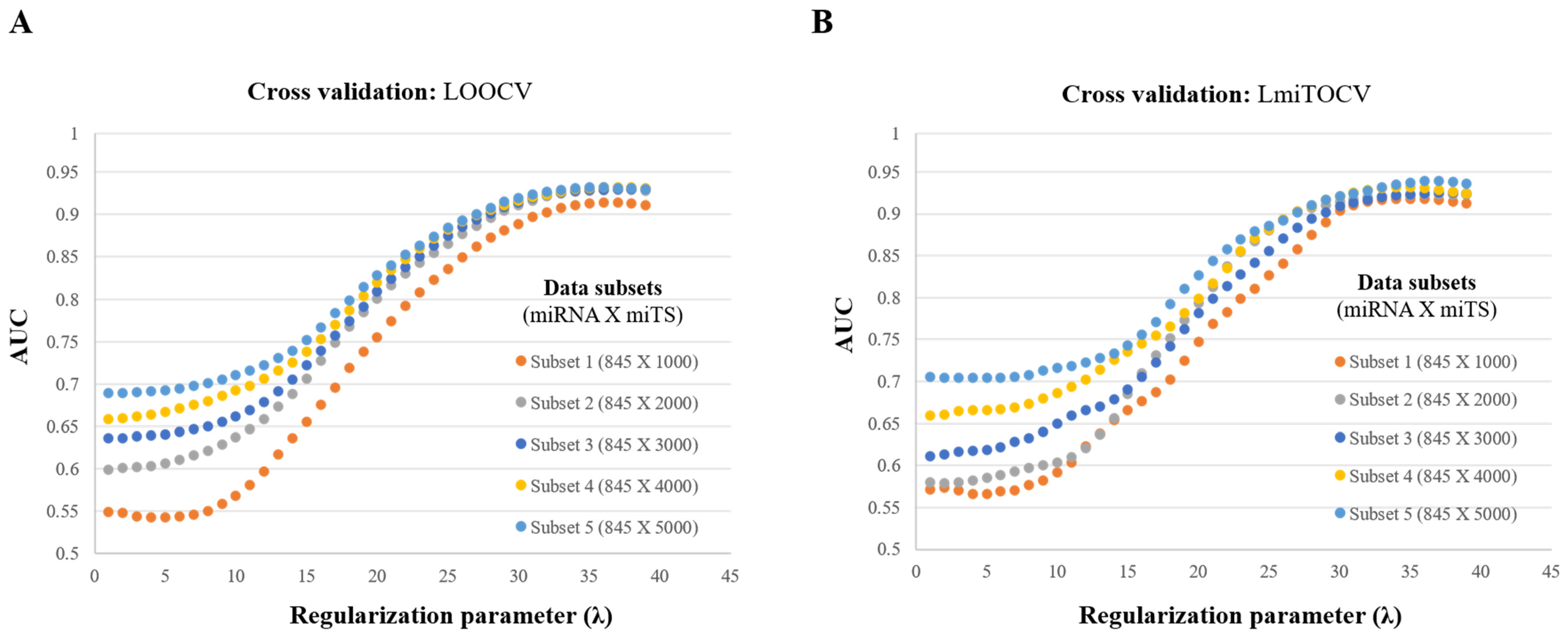
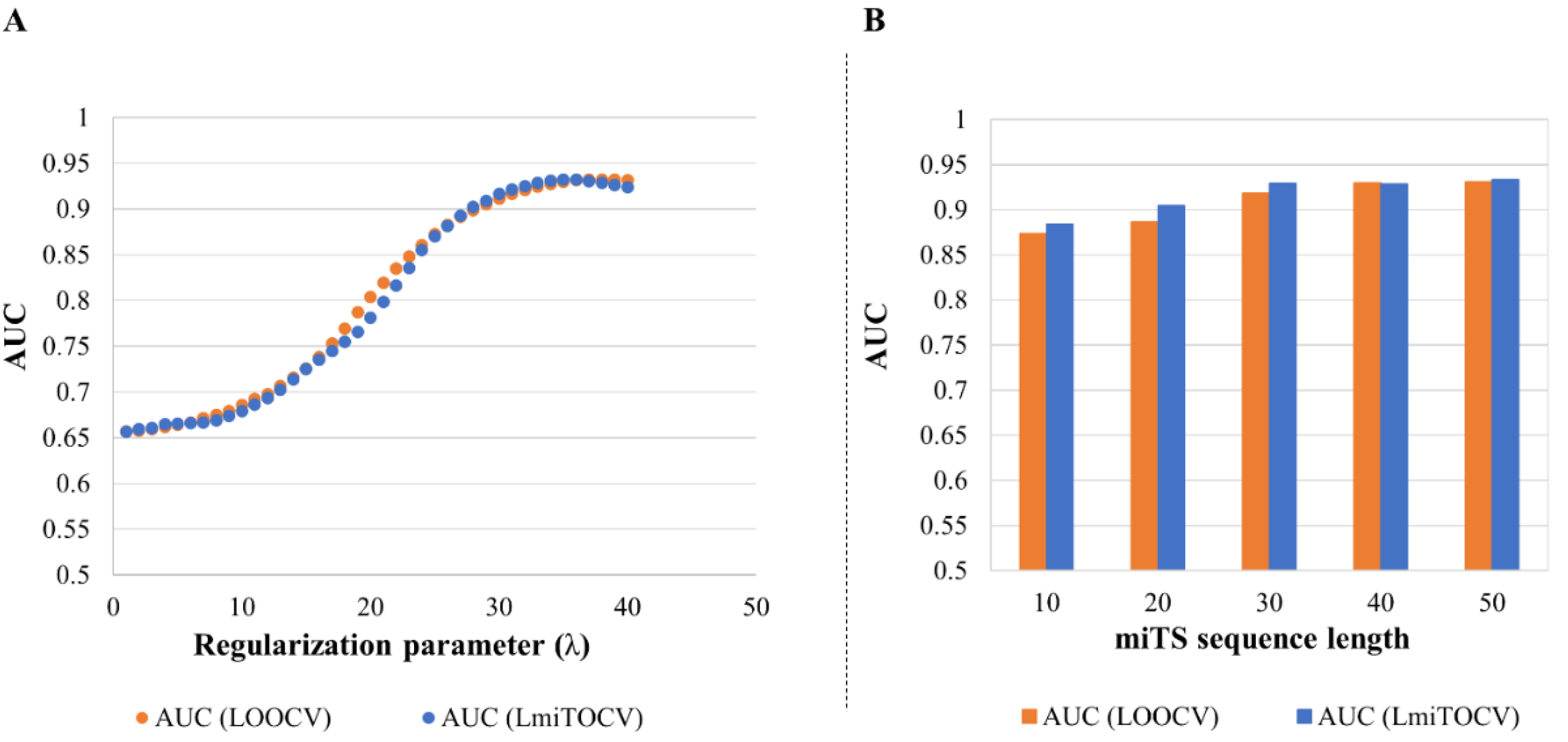
3.2. Evaluation of Regularization Parameter (λ)
3.3. Evaluation of miTS Sequence Length and Features
3.3.1. Effect of Longer Sequence Length
3.3.2. Feature Selection and Feature Contribution
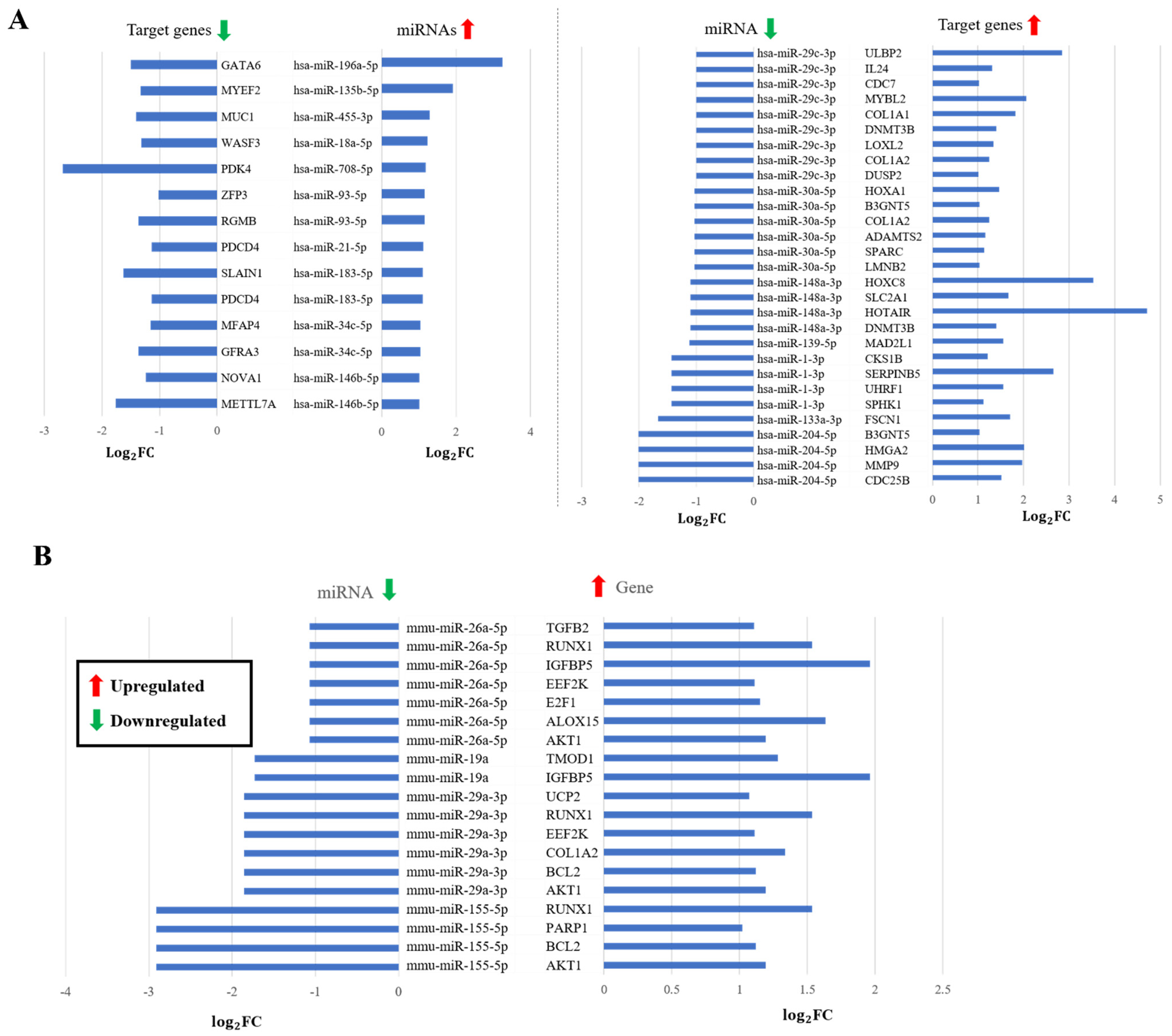
3.4. Validation
Supporting Predictions by Expression of miRNA and mRNA in Human and Mouse
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Methodology
Appendix A.1.1. miRNA/Gene Expression Analysis in Gastrointestinal (GI) Cancer
RNAseq Data Processing
miRNAseq Data Processing
miRNA Target Identification Using QIAGEN Ingenuity Pathway Analysis (IPA)

miRNA/Gene Expression Analysis in Control and Septic Mice
Appendix A.2. Calculation of Euclidean Distance Using Features
References
- Ivey, K.N.; Srivastava, D. microRNAs as developmental regulators. Cold Spring Harb. Perspect. Biol. 2015, 7, a008144. [Google Scholar] [CrossRef] [PubMed]
- Bär, C.; Thum, T.; De Gonzalo-Calvo, D. Circulating miRNAs as mediators in cell-to-cell communication. Epigenomics 2019, 11, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Harrandah, A.M.; Mora, R.A.; Chan, E.K.L. Emerging microRNAs in cancer diagnosis, progression, and immune surveillance. Cancer Lett. 2018, 438, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.S.; Tam, W.L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A Pattern-Based Method for the Identification of MicroRNA Binding Sites and Their Corresponding Heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.C.; Wani, S.; Steptoe, A.L.; Krishnan, K.; Nones, K.; Nourbakhsh, E.; Vlassov, A.; Grimmond, S.M.; Cloonan, N. Imperfect centered miRNA binding sites are common and can mediate repression of target mRNAs. Genome Biol. 2014, 15, R51. [Google Scholar] [CrossRef] [PubMed]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N. The mechanics of miRNA-mediated gene silencing: A look under the hood of miRISC. Nat. Struct. Mol. Biol. 2012, 19, 586–593. [Google Scholar] [CrossRef]
- Xu, W.; Lucas, A.S.; Wang, Z.; Liu, Y. Identifying microRNA targets in different gene regions. BMC Bioinform. 2014, 15, S4. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhou, W.; Liu, Y.; Liu, T.; Li, C.; Wang, L. Oncogenic role of microRNA-532-5p in human colorectal cancer via targeting of the 5′UTR of RUNX3. Oncol. Lett. 2018, 15, 7215–7220. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Sung, Y.M.; Park, J.; Kim, S.; Kim, J.; Park, J.; Ha, H.; Bae, J.Y.; Kim, S.; Baek, D. General rules for functional microRNA targeting. Nat. Genet. 2016, 48, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Rennie, W.A.; Carmack, C.S.; Kanoria, S.; Cheng, J.; Lu, J.; Ding, Y. Effects of genetic variations on microRNA: Target interactions. Nucleic Acids Res. 2014, 42, 9543–9552. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.M.; Thompson, J.A.; Ufkin, M.L.; Sathyanarayana, P.; Liaw, L.; Congdon, C.B. Common features of microRNA target prediction tools. Front. Genet. 2014, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. Mirwalk: An online resource for prediction of microrna binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar]
- Bandyopadhyay, S.; Ghosh, D.; Mitra, R.; Zhao, Z. MBSTAR: Multiple instance learning for predicting specific functional binding sites in microRNA targets. Sci. Rep. 2015, 5, 8004. [Google Scholar] [CrossRef]
- Wen, M.; Cong, P.; Zhang, Z.; Lu, H.; Li, T. DeepMirTar: A deep-learning approach for predicting human miRNA targets. Bioinformatics 2018, 34, 3781–3787. [Google Scholar] [CrossRef]
- Pla, A.; Zhong, X.; Rayner, S. miRAW: A deep learning-based approach to predict microRNA targets by analyzing whole microRNA transcripts. PLoS Comput. Biol. 2018, 14, e1006185. [Google Scholar] [CrossRef]
- Kyrollos, D.G.; Reid, B.; Dick, K.; Green, J.R. RPmirDIP: Reciprocal Perspective improves miRNA targeting prediction. Sci. Rep. 2020, 10, 11770. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.; Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015, 43, D146–D152. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, X. MiRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef]
- Kanoria, S.; Rennie, W.; Liu, C.; Carmack, C.S.; Lu, J.; Ding, Y. STarMir tools for prediction of microRNA binding sites. Methods Mol. Biol. 2016, 1490, 73–82. [Google Scholar] [PubMed]
- Vlachos, I.S.; Paraskevopoulou, M.D.; Karagkouni, D.; Georgakilas, G.; Vergoulis, T.; Kanellos, I.; Anastasopoulos, I.L.; Maniou, S.; Karathanou, K.; Kalfakakou, D.; et al. DIANA-TarBase v7.0: Indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015, 43, D153–D159. [Google Scholar] [CrossRef]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. MiRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef]
- Bottini, S.; Pratella, D.; Grandjean, V.; Repetto, E.; Trabucchi, M. Recent computational developments on CLIP-seq data analysis and microRNA targeting implications. Brief Bioinform. 2017, 19, 1290–1301. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y. Current experimental strategies for intracellular target identification of microRNA. ExRNA 2019, 62, 129–137. [Google Scholar] [CrossRef]
- Schäfer, M.; Ciaudo, C. Prediction of the miRNA interactome—Established methods and upcoming perspectives. Comput. Struct. Biotechnol. J. 2020, 18, 548–557. [Google Scholar] [CrossRef]
- Gerlach, W.; Giegerich, R. GUUGle: A utility for fast exact matching under RNA complementary rules including G-U base pairing. Bioinformatics 2006, 22, 762–764. [Google Scholar] [CrossRef]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human microRNA targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef] [Green Version]
- Riolo, G.; Cantara, S.; Marzocchi, C.; Ricci, C. miRNA targets: From prediction tools to experimental validation. Methods Protoc. 2021, 4, 1. [Google Scholar] [CrossRef]
- Jiang, H.; Yang, M.; Chen, X.; Li, M.; Li, Y.; Wang, J. MiRTMC: A miRNA Target Prediction Method Based on Matrix Completion Algorithm. IEEE J. Biomed. Health Inform. 2020, 24, 3630–3641. [Google Scholar] [CrossRef]
- Parveen, A.; Mustafa, S.H.; Yadav, P.; Kumar, A. Applications of Machine Learning in miRNA Discovery and Target Prediction. Curr. Genom. 2020, 20, 537–544. [Google Scholar] [CrossRef]
- Plotnikova, O.M.; Skoblov, M.Y. Efficiency of the miRNA- mRNA Interaction Prediction Programs. Mol. Biol. 2018, 52, 543–554. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, L.; Li, X.; Zhang, Y.; Xu, S.; Huang, X. Prediction of miRNA targets by learning from interaction sequences. PLoS ONE 2020, 15, e0232578. [Google Scholar] [CrossRef]
- Long, D.; Lee, R.; Williams, P.; Chan, C.Y.; Ambros, V.; Ding, Y. Potent effect of target structure on microRNA function. Nat. Struct. Mol. Biol. 2007, 14, 287–294. [Google Scholar] [CrossRef]
- Fiannaca, A.; La Rosa, M.; La Paglia, L.; Rizzo, R.; Urso, A. MiRNATIP: A SOM-based miRNA-target interactions predictor. BMC Bioinform. 2016, 17, 321. [Google Scholar] [CrossRef]
- Ghoshal, A.; Shankar, R.; Bagchi, S.; Grama, A.; Chaterji, S. MicroRNA target prediction using thermodynamic and sequence curves. BMC Genom. 2015, 16, 999. [Google Scholar]
- Krüger, J.; Rehmsmeier, M. RNAhybrid: MicroRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef]
- Robins, H.; Li, Y.; Padgett, R.W. Incorporating structure to predict microRNA targets. Proc. Natl. Acad. Sci. USA 2005, 102, 4006–4009. [Google Scholar] [CrossRef]
- van Laarhoven, T.; Marchiori, E. Predicting Drug-Target Interactions for New Drug Compounds Using a Weighted Nearest Neighbor Profile. PLoS ONE 2013, 27, 805–828. [Google Scholar]
- Yan, C.; Wang, J.; Lan, W.; Wu, F.X.; Pan, Y. SDTRLS: Predicting Drug-Target Interactions for Complex Diseases Based on Chemical Substructures. Complexity 2017, 2017, 2713280. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Duan, G.; Pan, Y.; Wu, F.X.; Wang, J. DDIGIP: Predicting drug-drug interactions based on Gaussian interaction profile kernels. BMC Bioinform. 2019, 20, 538. [Google Scholar] [CrossRef]
- Yan, C.; Wang, J.; Ni, P.; Lan, W.; Wu, F.X.; Pan, Y. DNRLMF-MDA:Predicting microRNA-Disease Associations Based on Similarities of microRNAs and Diseases. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 233–243. [Google Scholar] [CrossRef]
- Chen, X.; Yan, G.Y. Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics 2013, 29, 2617–2624. [Google Scholar] [CrossRef]
- Kehl, T.; Backes, C.; Kern, F.; Fehlmann, T.; Ludwig, N.; Meese, E.; Lenhof, H.P.; Keller, A. About miRNAs, miRNA seeds, target genes and target pathways. Oncotarget 2017, 8, 107167–107175. [Google Scholar] [CrossRef]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Witkos, T.M.; Krzyzosiak, W.J.; Fiszer, A.; Koscianska, E. A potential role of extended simple sequence repeats in competing endogenous RNA crosstalk. RNA Biol. 2018, 15, 1399–1409. [Google Scholar] [CrossRef]
- Zheng, Z.; Reichel, M.; Deveson, I.; Wong, G.; Li, J.; Millar, A.A. Target RNA Secondary Structure Is a Major Determinant of miR159 Efficacy. Plant Physiol. 2017, 174, 1764–1778. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 2019, 20, 18. [Google Scholar] [CrossRef]
- Pahikkala, T.; Airola, A.; Pietilä, S.; Shakyawar, S.; Szwajda, A.; Tang, J.; Aittokallio, T. Toward more realistic drug-target interaction predictions. Brief Bioinform. 2014, 16, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Kimeldorf, G.; Wahba, G. Some results on Tchebycheffian spline functions. J Math. Anal. Appl. 1971, 33, 82–95. [Google Scholar] [CrossRef] [Green Version]
- Hue, M.; Riffle, M.; Vert, J.P.; Noble, W.S. Large-scale prediction of protein-protein interactions from structures. BMC Bioinform. 2010, 11, 144. [Google Scholar] [CrossRef]
- Rifkin, R.; Yeo, G.; Poggio, T. Regularized Least-Squares Classification. Nato Sci. Ser. Sub Ser. III Comput. Syst. Sci. 2003, 190, 131–153. [Google Scholar]
- van Laarhoven, T.; Nabuurs, S.B.; Marchiori, E. Gaussian interaction profile kernels for predicting drug-target interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar] [CrossRef]
- Luo, J.; Xiao, Q.; Liang, C.; DIng, P. Predicting MicroRNA-Disease Associations Using Kronecker Regularized Least Squares Based on Heterogeneous Omics Data. IEEE Access 2017, 5, 2503–2513. [Google Scholar] [CrossRef]
- Tokar, T.; Pastrello, C.; Rossos, A.E.M.; Abovsky, M.; Hauschild, A.C.; Tsay, M.; Lu, R.; Jurisica, I. MirDIP 4.1—Integrative database of human microRNA target predictions. Nucleic Acids Res. 2018, 46, D360–D370. [Google Scholar]
- Rooda, I.; Hensen, K.; Kaselt, B.; Kasvandik, S.; Pook, M.; Kurg, A.; Salumets, A.; Velthut-Meikas, A. Target prediction and validation of microRNAs expressed from FSHR and aromatase genes in human ovarian granulosa cells. Sci. Rep. 2020, 10, 2300. [Google Scholar] [CrossRef]
- Kern, F.; Krammes, L.; Danz, K.; Diener, C.; Kehl, T.; Küchler, O.; Fehlmann, T.; Kahraman, M.; Rheinheimer, S.; Aparicio-Puerta, E.; et al. Validation of human microRNA target pathways enables evaluation of target prediction tools. Nucleic Acids Res. 2021, 49, 127–144. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, D. The pattern of microRNA binding site distribution. Genes 2017, 8, 296. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Yu, H. DR2DI: A powerful computational tool for predicting novel drug-disease associations. J. Comput. Aided Mol. Des. 2018, 32, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Smita, S.; Mishra, D.C.; Kumar, S.; Singh, B.K.; Rai, A. Abiotic stress responsive mirna-target network and related markers (SNP, SSR) in Brassica juncea. Front. Plant Sci. 2017, 8, 1943. [Google Scholar] [CrossRef]
- Patil, P.G.; Singh, N.V.; Parashuram, S.; Bohra, A.; Mundewadikar, D.M.; Sangnure, V.R.; Babu, K.D.; Sharma, J. Genome wide identification, characterization and validation of novel miRNA-based SSR markers in pomegranate (Punica granatum L.). Physiol. Mol. Biol. Plants 2020, 26, 683–696. [Google Scholar]
- Riffo-Campos, Á.L.; Riquelme, I.; Brebi-Mieville, P. Tools for sequence-based miRNA target prediction: What to choose? Int. J. Mol. Sci. 2016, 17, 1987. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Ziebarth, J.D.; Cui, Y. PolymiRTS Database 3.0: Linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Res. 2014, 42, D86–D91. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Badal, S.S.; Wang, Y.; Chang, B.H.J.; Rodriguez, A.; Danesh, F.R. MicroRNA-22 is a master regulator of bone morphogenetic protein-7/6 homeostasis in the kidney. J. Biol. Chem. 2013, 288, 36202–36214. [Google Scholar] [CrossRef]
- Garo, L.P.; Ajay, A.K.; Fujiwara, M.; Gabriely, G.; Raheja, R.; Kuhn, C.; Kenyon, B.; Skillin, N.; Kadowaki-Saga, R.; Saxena, S.; et al. MicroRNA-146a limits tumorigenic inflammation in colorectal cancer. Nat. Commun. 2021, 12, 2419. [Google Scholar] [CrossRef]
- Chaudhuri, E.; Dash, S.; Balasubramaniam, M.; Padron, A.; Holland, J.; Sowd, G.A.; Villalta, F.; Engelman, A.N.; Pandhare, J.; Dash, C. The HIV-1 capsid-binding host factor CPSF6 is posttranscriptionally regulated by the cellular microRNA miR-125b. J. Biol. Chem. 2020, 295, 5081–5094. [Google Scholar] [CrossRef]
- Dash, S.; Dash, C.; Pandhare, J. Therapeutic significance of microRNA-mediated regulation of PARP-1 in SARS-CoV-2 infection. Non-Coding RNA 2021, 7, 60. [Google Scholar] [CrossRef]
- Dash, S.; Balasubramaniam, M.; Dash, C.; Pandhare, J. Biotin-based pulldown assay to validate mRNA targets of cellular miRNAs. J. Vis. Exp. 2018, 136, 57786. [Google Scholar] [CrossRef] [PubMed]
- Theil, K.; Imami, K.; Rajewsky, N. Identification of proteins and miRNAs that specifically bind an mRNA in vivo. Nat. Commun. 2019, 10, 4205. [Google Scholar] [PubMed]
- Dash, S.; Balasubramaniam, M.; Martínez-Rivera, F.J.; Godino, A.; Peck, E.G.; Patnaik, S.; Suar, M.; Calipari, E.S.; Nestler, E.J.; Villalta, F.; et al. Cocaine-regulated microRNA miR-124 controls poly (ADP-ribose) polymerase-1 expression in neuronal cells. Sci. Rep. 2020, 10, 11197. [Google Scholar] [PubMed]
- Hunter, S.E.; Finnegan, E.F.; Zisoulis, D.G.; Lovci, M.T.; Melnik-Martinez, K.V.; Yeo, G.W.; Pasquinelli, A.E. Functional Genomic Analysis of the let-7 Regulatory Network in Caenorhabditis elegans. PLoS Genet. 2013, 9, e1003353. [Google Scholar] [CrossRef] [PubMed]
- Torres Fernández, L.A.; Mitschka, S.; Ulas, T.; Weise, S.; Dahm, K.; Becker, M.; Händler, K.; Beyer, M.; Windhausen, J.; Schultze, J.L.; et al. The stem cell-specific protein TRIM71 inhibits maturation and activity of the pro-differentiation miRNA let-7 via two independent molecular mechanisms. RNA 2021, 27, 805–828. [Google Scholar] [CrossRef]
- Hyeon, H.K.; Kuwano, Y.; Srikantan, S.; Eun, K.L.; Martindale, J.L.; Gorospe, M. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 2009, 23, 1743–1748. [Google Scholar]
- Fan, X.; Kurgan, L. Comprehensive overview and assessment of computational prediction of microRNA targets in animals. Brief. Bioinform. 2014, 17, 321. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.J.; Maticzka, D.; Moḧl, M.; Gagnon, J.N.; Brown, C.M.; Backofen, R. Global or local? Predicting secondary structure and accessibility in mRNAs. Nucleic Acids Res. 2012, 16, 780–794. [Google Scholar] [CrossRef]
- Garcia, D.M.; Baek, D.; Shin, C.; Bell, G.W.; Grimson, A.; Bartel, D.P. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat. Struct. Mol. Biol. 2010, 18, 1139–1146. [Google Scholar]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accuracy | Sensitivity | Specificity | MCC | AUC (ROC Curve) | |
|---|---|---|---|---|---|
| Human dataset | |||||
| LOOCV | 0.908 | 0.847 | 0.909 | 0.67 | 0.931 |
| LmiTOCV | 0.91 | 0.829 | 0.909 | 0.652 | 0.925 |
| Mouse dataset | |||||
| LOOCV | 0.846 | 0.783 | 0.846 | 0.59 | 0.861 |
| LmiTOCV | 0.844 | 0.767 | 0.839 | 0.564 | 0.863 |
| miRNA | Target Gene | Results in Reference | mintRULS | Experimental Evidence | ||
|---|---|---|---|---|---|---|
| Predictions (Quartile) | Classification | Cells/Tissues | Reference | |||
| hsa-miR-548ba | LIFR | Target | Upper | Strong Target | ovarian granulosa cells | [59] |
| PTEN | Target | Upper | Strong Target | |||
| NEO1 | Target | Upper | Strong Target | |||
| hsa-miR-34a-5p | CLOCK | Target | Upper | Strong Target | SH-SY5Y cells | [60] |
| CREB1 | Target | Upper | Strong Target | |||
| GRIA4 | Target | Lower | Weak Target | |||
| SMAD2 | Target | Upper | Strong Target | |||
| SMAD7 | Target | Upper | Strong Target | |||
| hsa-miR-22 | BMP-7/6 | Target | Upper | Strong Target | Mouse primary kidney fibroblasts | [67] |
| hsa-miR-146a-3p | TRAF6 | Target | Upper | Strong Target | Mouse Myeloid cells | [68] |
| RIPK2 | Target | Upper | Strong Target | |||
| hsa-miR-125b | CPSF6 | Target | Upper | Strong Target | HEK-293T | [69] |
| PARP1 | Target | Middle | Moderate Target | HEK-293T cells | [70,71] | |
| p53 | Target | Upper | Strong Target | |||
| Beta-actin | Non-Target | Lower | Weak Target | |||
| 18S RNA | gld-1:gfp | Non-Target | Lower | Weak Target | Caenorhabditis elegans | [72] |
| miRNA | miRNA/Seed Mutation | Target Gene/Mutation | Result in Reference | mintRULS Prediction | Reference | |
|---|---|---|---|---|---|---|
| Quartile | Class | |||||
| hsa-miR-124-3p | UAAGGCACGCGGUGAAUGCCAA | Parp-1 (WT) | Target | Upper | Strong Target | [73] |
| Mut1: PARP-1 (CC > GG) | No target | Lower | Weak Target | |||
| Mut2: PARP-1 (TG > CA) | No target | Lower | Weak Target | |||
| Mut3: PARP-1 (GC > AA) | No target | Lower | Weak Target | |||
| Mut4: deletion (ΔGC) | No target | Middle | Moderate Target | |||
| cel-let-7-3p | AU[G/A]CAA | LIN-41 | WT: Target | Upper | Strong Target | [74] |
| Mutation: No Target | Lower * | Weak Target * | ||||
| hsa-miR-662 | CCCAC[G/A]U | KLLN | Disrupted (∆S = −0.51) | Upper | Strong Target | PolymiRTS database |
| Lower * | Weak Target * | |||||
| PATE4 | Disrupted (∆S = −0.45) | Upper | Strong Target | PolymiRTS database | ||
| Lower * | Weak Target * | |||||
| hsa-miR-125a-5p | CCCUGA[G/U] | ZMYM3 | Disrupted (∆S = −0.31) | Upper | Strong Target | PolymiRTS database |
| Lower * | Moderate Target * | |||||
| PRRC1 | Disrupted (∆S = −0.45) | Upper | Strong Target | PolymiRTS database | ||
| Lower * | Weak Target * | |||||
| AQPEP | Disrupted (∆S = −0.42) | Upper | Strong Target | PolymiRTS database | ||
| Lower * | Weak Target * | |||||
| hsa-miR-645 | [C/G]UAGGCU | COL4A4 | Disrupted (∆S = −0.38) | Upper | Strong Target | PolymiRTS database |
| Middle * | Moderate Target * | |||||
| MAOA | Disrupted (∆S = −0.4) | Upper | Strong Target | PolymiRTS database | ||
| Lower * | Weak Target * | |||||
| IL4R | Disrupted (∆S = −0.42) | Upper | Strong Target | PolymiRTS database | ||
| Lower * | Weak Target * | |||||
| hsa-miR-146a-3p | CP | Disrupted (∆S = −0.57) | Upper | Strong Target | PolymiRTS database | |
| Lower * | Weak Target * | |||||
| ABCB1 | Disrupted (∆S = −0.35) | Upper | Strong Target | PolymiRTS database | ||
| Lower * | Weak Target * | |||||
| mmu-miR-342-5p | [G/-]GGGUGC | PIGU | Disrupted (∆S = −0.46) | Upper | Strong Target | PolymiRTS database |
| Lower * | Weak Target * | |||||
| RASL10B | Disrupted (∆S = −0.5) | Middle | Moderate Target | PolymiRTS database | ||
| Lower * | Weak Target * | |||||
| MCU | Disrupted (∆S = −0.54) | Upper | Strong Target | PolymiRTS database | ||
| Lower * | Weak Target * | |||||
| mmu-miR-690 | AAGGCU[A/G] | CNOT6 | Disrupted (∆S = −0.3) | Upper | Strong Target | PolymiRTS database |
| Lower * | Weak Target * | |||||
| ELOVL4 | Disrupted (∆S = −0.35) | Upper | Strong Target | PolymiRTS database | ||
| Lower * | Weak Target * | |||||
| RBBP5 | Disrupted (∆S = −0.34) | Upper | Strong Target | PolymiRTS database | ||
| Middle * | Moderate Target * | |||||
| mmu-miR-743a-3p | AAAGAC[A/G] | MXI1 | Disrupted (∆S = −0.33) | Upper | Strong Target | PolymiRTS database |
| Lower * | Weak Target * | |||||
| PRRG3 | Disrupted (∆S = −0.51) | Upper | Strong Target | PolymiRTS database | ||
| Lower * | Weak Target * | |||||
| MBNL3 | Disrupted (∆S = −0.43) | Upper | Strong Target | PolymiRTS database | ||
| Lower * | Weak Target * | |||||
| miRNA | Target Gene | mintRULS | Evidence (Literature/Databases) | |
|---|---|---|---|---|
| Prediction Class (Quartile) | Classification | |||
| hsa-miR-3941 | TNPO1 | Upper | Strong Target | miRDB |
| hsa-let-7d-5p | BACH1 | Upper | Strong Target | TargetScan |
| hsa-let-7d-5p | BCL2L1 | Upper | Strong Target | TargetScan |
| hsa-let-7d-5p | NCAM1 | Upper | Strong Target | New |
| hsa-let-7d-5p | TIMP3 | Upper | Strong Target | New |
| hsa-let-7d-5p | IL6R | Upper | Strong Target | TargetScan, miRDB |
| hsa-let-7d-5p | CD44 | Upper | Strong Target | New |
| hsa-let-7d-5p | ITGB3 | Upper | Strong Target | TargetScan, miRDB |
| hsa-let-7d-5p | CCNE1 | Upper | Strong Target | miRDB |
| hsa-let-7d-5p | MAP4K3 | Upper | Strong Target | TargetScan |
| hsa-let-7d-5p | PTEN | Upper | Strong Target | New |
| hsa-let-7e-5p | TRIM71 | Upper | Strong Target | TargetScan, [75] |
| hsa-let-7e-5p | ZBTB7A | Upper | Strong Target | New |
| hsa-let-7e-5p | KLF9 | Upper | Strong Target | TargetScan |
| hsa-let-7e-5p | IGFBP5 | Upper | Strong Target | New |
| hsa-let-7e-5p | ALDH5A1 | Upper | Strong Target | New |
| hsa-let-7e-5p | CDK4 | Upper | Strong Target | New |
| hsa-let-7e-5p | BCL2L1 | Upper | Strong Target | miRDB |
| hsa-let-7e-5p | MDM4 | Upper | Strong Target | TargetScan |
| hsa-let-7e-5p | TIMP3 | Upper | Strong Target | [76] |
| hsa-let-7e-5p | PAPPA | Middle | Moderate Target | TargetScan |
| hsa-let-7e-5p | MYC | Upper | Strong Target | [76] |
| hsa-miR-106b-5p | NLN | Upper | Strong Target | TargetScan |
| hsa-miR-106b-5p | SLC6A4 | Upper | Strong Target | TargetScan |
| hsa-miR-106b-5p | GPD2 | Upper | Strong Target | TargetScan |
| hsa-miR-106b-5p | RASA1 | Upper | Strong Target | TargetScan |
| hsa-miR-106b-5p | EGLN1 | Upper | Strong Target | TargetScan |
| hsa-miR-106b-5p | ATAT1 | Upper | Strong Target | New |
| hsa-miR-106b-5p | PAX6 | Upper | Strong Target | miRDB |
| hsa-miR-106b-5p | PBX3 | Upper | Strong Target | TargetScan |
| hsa-miR-106b-5p | MCL1 | Upper | Strong Target | TargetScan |
| hsa-miR-106b-5p | FLT1 | Middle | Moderate Target | TargetScan miRDB |
| hsa-miR-106b-5p | FXN | Middle | Moderate Target | miRDB |
| Cancer Type | Expression | IPA | mintRULS | ||||||
|---|---|---|---|---|---|---|---|---|---|
| miRNA | Target Gene | Exp. Observed* | High Predicted | Total | Strong-Target | Moderate-Target | Weak-Target | Total | |
| STAD |  |  | 13 | 77 | 90 | 28 | 46 | 16 | 90 |
 |  | 15 | 11 | 26 | 16 | 9 | 1 | 26 | |
| CHOL |  |  | 21 | 134 | 155 | 71 | 64 | 20 | 155 |
 |  | 80 | 169 | 249 | 125 | 101 | 23 | 249 | |
| ESCA |  |  | 36 | 20 | 56 | 29 | 21 | 6 | 56 |
 |  | 4 | 20 | 24 | 14 | 8 | 2 | 24 | |
| LIHC |  |  | 3 | 4 | 7 | 7 | 0 | 0 | 7 |
 |  | 23 | 19 | 42 | 42 | 0 | 0 | 42 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shakyawar, S.; Southekal, S.; Guda, C. mintRULS: Prediction of miRNA–mRNA Target Site Interactions Using Regularized Least Square Method. Genes 2022, 13, 1528. https://doi.org/10.3390/genes13091528
Shakyawar S, Southekal S, Guda C. mintRULS: Prediction of miRNA–mRNA Target Site Interactions Using Regularized Least Square Method. Genes. 2022; 13(9):1528. https://doi.org/10.3390/genes13091528
Chicago/Turabian StyleShakyawar, Sushil, Siddesh Southekal, and Chittibabu Guda. 2022. "mintRULS: Prediction of miRNA–mRNA Target Site Interactions Using Regularized Least Square Method" Genes 13, no. 9: 1528. https://doi.org/10.3390/genes13091528
APA StyleShakyawar, S., Southekal, S., & Guda, C. (2022). mintRULS: Prediction of miRNA–mRNA Target Site Interactions Using Regularized Least Square Method. Genes, 13(9), 1528. https://doi.org/10.3390/genes13091528