
Identification of Differential Expression Genes between Volume and Pressure Overloaded Hearts Based on Bioinformatics Analysis

 , , and
, , and
Abstract
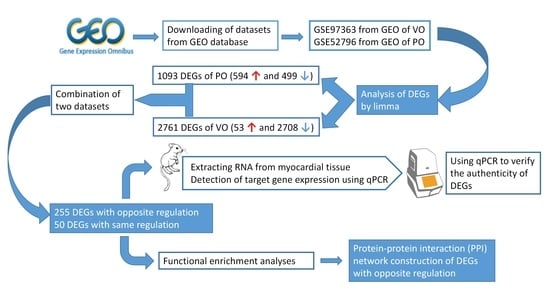
:
1. Introduction
2. Materials and Methods
2.1. Microarray Data Collection and Preprocessing
2.2. Study Design and Differentially Expressed Gene Screening
2.3. Functional Enrichment Analysis
2.4. Protein–Protein Interaction (PPI) Network Construction
2.5. Mice and Surgery
2.6. RNA Isolation and Quantitative PCR Analysis
3. Results
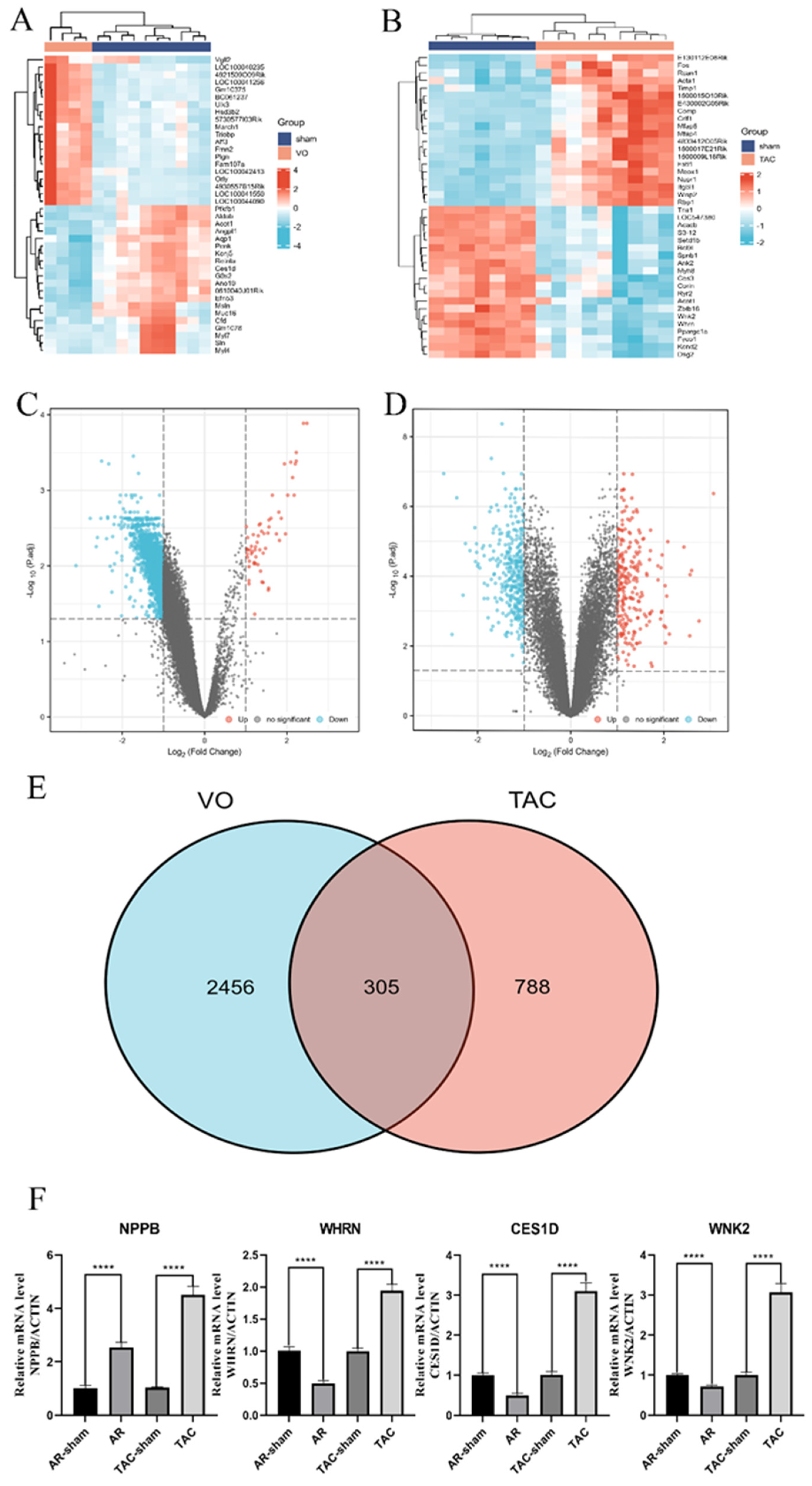
3.1. Identification of DEGs and Verification of qRT-PCR
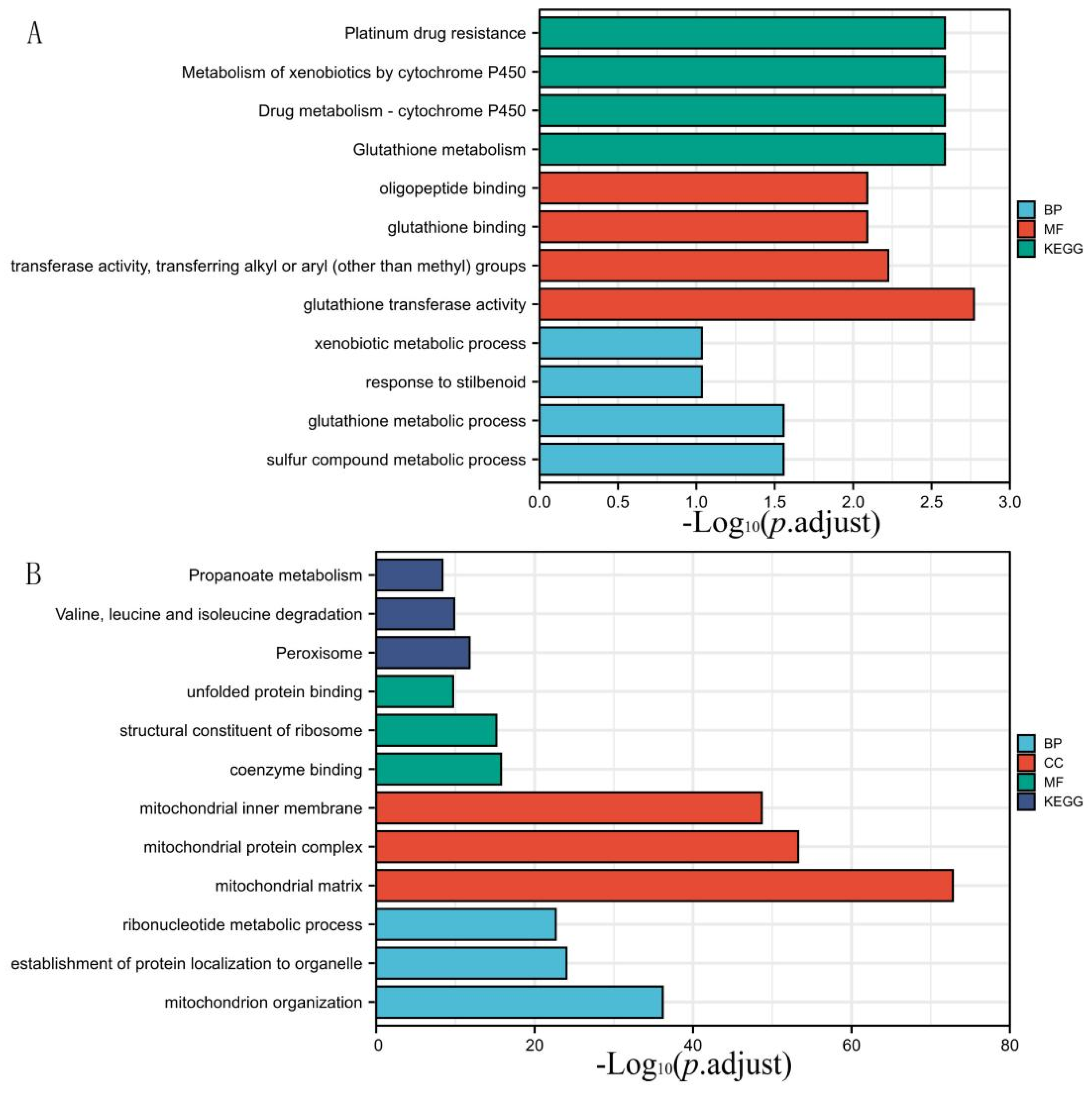
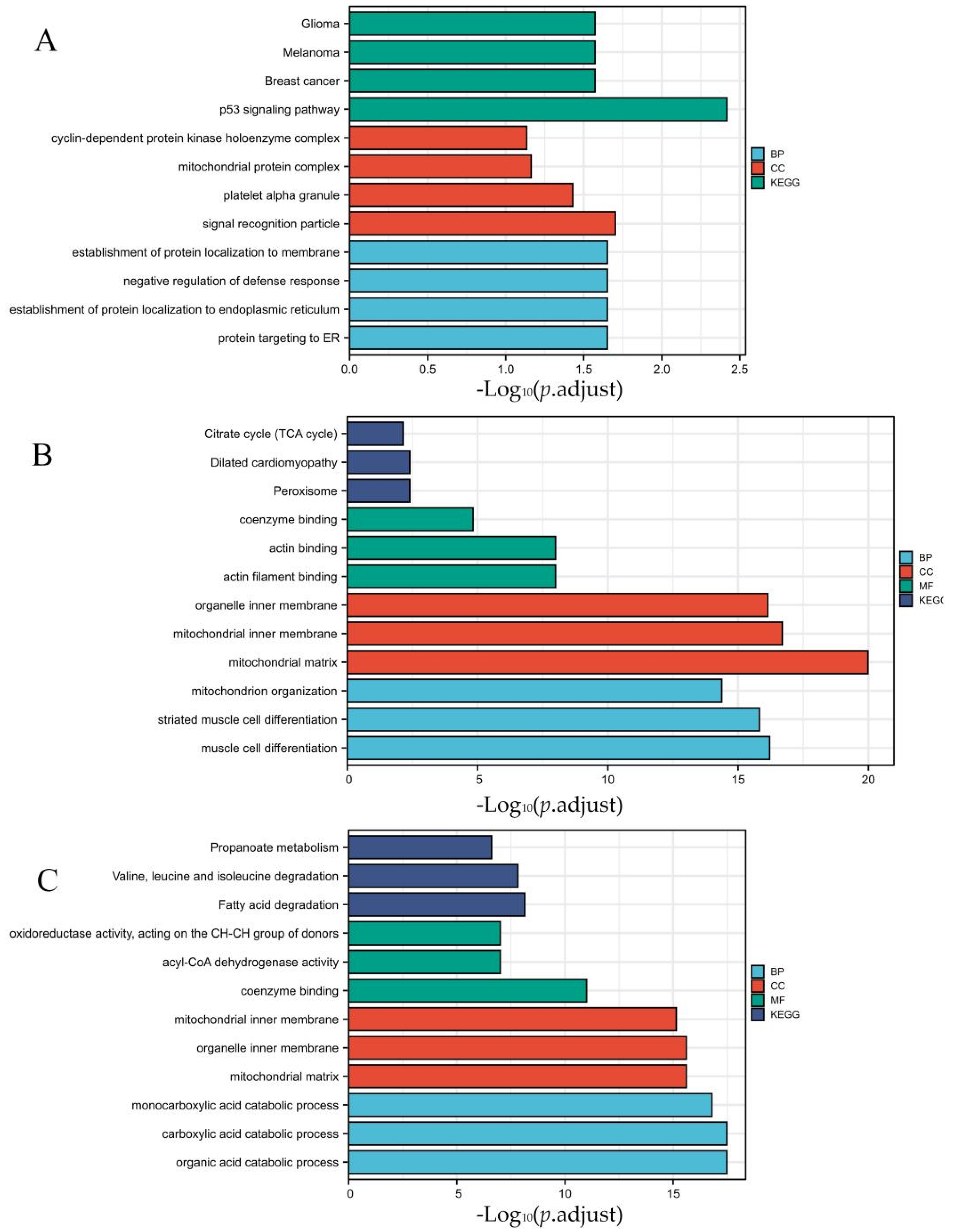
3.2. DEGs Co-Expression Results and Functional Enrichment Analysis
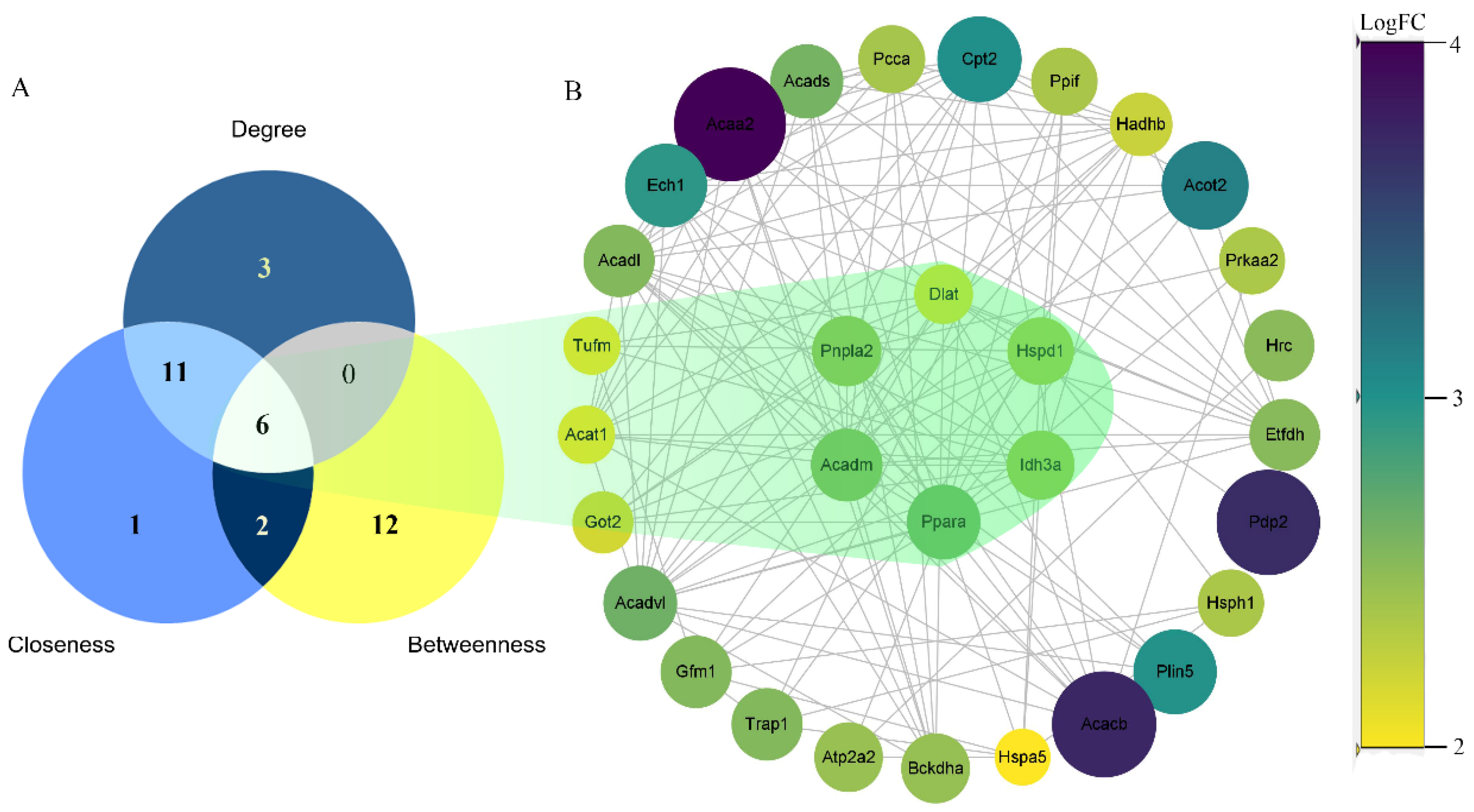
3.3. Protein–Protein Interaction (PPI) Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mosterd, A.; Hoes, A.W. Clinical epidemiology of heart failure. Heart 2007, 93, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Metra, M.; Teerlink, J.R. Heart failure. Lancet 2017, 390, 1981–1995. [Google Scholar] [CrossRef]
- Schwinger, R. Pathophysiology of heart failure. Cardiovasc. Diagn. Ther. 2021, 11, 263–276. [Google Scholar] [CrossRef]
- Klein, I.; Danzi, S. Thyroid Disease and the Heart. Curr. Probl. Cardiol. 2016, 41, 65–92. [Google Scholar] [CrossRef] [Green Version]
- Osuna, P.M.; Udovcic, M.; Sharma, M.D. Hyperthyroidism and the Heart. Methodist DeBakey Cardiovasc. J. 2017, 13, 60–63. [Google Scholar] [CrossRef] [Green Version]
- Limongelli, G.; Rubino, M.; Esposito, A.; Russo, M.; Pacileo, G. The challenge of cardiomyopathies and heart failure in pregnancy. Curr. Opin. Obstet. Gynecol. 2018, 30, 378–384. [Google Scholar] [CrossRef]
- Rangaswami, J.; McCullough, P.A. Heart Failure in End-Stage Kidney Disease: Pathophysiology, Diagnosis, and Therapeutic Strategies. Semin. Nephrol. 2018, 38, 600–617. [Google Scholar] [CrossRef]
- Sîrbu, O.; Floria, M.; Dascalita, P.; Victorita, S.; Laurentiu, S. Anemia in heart failure-from guidelines to controversies and challenges. Anatol. J. Cardiol. 2018, 20, 52–59. [Google Scholar] [CrossRef]
- Vonck, S.; Lanssens, D.; Staelens, A.S.; Kathleen, T.; Jolien, O.; Liesbeth, B.; Wilfried, G. Obesity in pregnancy causes a volume overload in third trimester. Eur. J. Clin. Investig. 2019, 49, e13173. [Google Scholar] [CrossRef]
- Simmonds, S.J.; Cuijpers, I.; Heymans, S.; Jones, E. Cellular and Molecular Differences between HFpEF and HFrEF: A Step Ahead in an Improved Pathological Understanding. Cells 2020, 9, 242. [Google Scholar] [CrossRef] [Green Version]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Héctor, B.; John, G.F.; Andrew, J.S.; Volkmar, F.; José, R.; Veli-Pekka, H.; Ewa, A.J.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar]
- Katz, S.D.; Hryniewicz, K.; Hriljac, I.; Kujtim, B.; Clarito, D.; Alhakam, H.; Aleksandr, Y. Vascular endothelial dysfunction and mortality risk in patients with chronic heart failure. Circulation 2005, 111, 310–314. [Google Scholar] [CrossRef] [Green Version]
- Penn, M.S. The role of leukocyte-generated oxidants in left ventricular remodeling. Am. J. Cardiol. 2008, 101, 30D–33D. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [Green Version]
- Zile, M.R.; Gottdiener, J.S.; Hetzel, S.J.; John, J.M.; Michel, K.; Robert, M.; Catalin, F.B.; Barry, M.M.; Peter, E.C. Prevalence and significance of alterations in cardiac structure and function in patients with heart failure and a preserved ejection fraction. Circulation 2011, 124, 2491–2501. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Jill, T.K.; Michelle, L.D.; Ryan, D.; Anjana, C.; Lina, W.; Komal, A.; Nadia, A.R.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Van Linthout, S.; Tschöpe, C. Inflammation-Cause or Consequence of Heart Failure or Both. Curr. Heart Fail. Rep. 2017, 14, 251–265. [Google Scholar] [CrossRef] [Green Version]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef]
- Raya, T.E.; Gay, R.G.; Lancaster, L.; Aguirre, M.; Moffett, C.; Goldman, S. Serial changes in left ventricular relaxation and chamber stiffness after large myocardial infarction in rats. Circulation 1988, 77, 1424–1431. [Google Scholar] [CrossRef] [Green Version]
- Stoddard, M.F.; Pearson, A.C.; Kern, M.J.; Ratcliff, J.; Mrosek, D.G.; Labovitz, A.J. Left ventricular diastolic function: Comparison of pulsed Doppler echocardiographic and hemodynamic indexes in subjects with and without coronary artery disease. J. Am. Coll. Cardiol. 1989, 13, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Aoyagi, T.; Pouleur, H.; Van Eyll, C.; Rousseau, M.F.; Mirsky, I. Wall motion asynchrony is a major determinant of impaired left ventricular filling in patients with healed myocardial infarction. Am. J. Cardiol. 1993, 72, 268–272. [Google Scholar] [CrossRef]
- Nakao, M.; Shimizu, I.; Katsuumi, G.; Yohko, Y.; Masayoshi, S.; Yuka, H.; Ryutaro, I.; Yung, T.H.; Shujiro, O.; Tomoyoshi, S.; et al. Empagliflozin maintains capillarization and improves cardiac function in a murine model of left ventricular pressure overload. Sci. Rep. 2021, 11, 18384. [Google Scholar] [CrossRef]
- Sun, S.; Hu, Y.; Xiao, Y.; Wang, S.; Jiang, C.; Liu, J.; Zhang, H.; Hong, H.; Li, F.; Ye, L. Postnatal Right Ventricular Developmental Track Changed by Volume Overload. J. Am. Heart Assoc. 2021, 10, e020854. [Google Scholar] [CrossRef]
- Toischer, K.; Zhu, W.; Hünlich, M.; Belal, A.M.; Sara, K.; Sean, P.R.; Katrin, S.; Deepak, R.; Stefan, E.; Loren, J.F.; et al. Cardiomyocyte proliferation prevents failure in pressure overload but not volume overload. J. Clin. Investig. 2017, 127, 4285–4296. [Google Scholar] [CrossRef] [Green Version]
- Mayer, S.C.; Gilsbach, R.; Preissl, S.; Elsa, B.M.; Tilman, S.; Nadine, B.; Achim, L.; Carolin, R.; Hannah, I.; Heiko, B.; et al. Adrenergic Repression of the Epigenetic Reader MeCP2 Facilitates Cardiac Adaptation in Chronic Heart Failure. Circ. Res. 2015, 117, 622–633. [Google Scholar] [CrossRef]
- Reddy, S.; Hu, D.Q.; Zhao, M.; Eddie, B.J.; Nefthi, S.; Ong, S.G.; Gwanghyun, J.; Kristina, B.K.; Michael, C.; Giovanni, F.; et al. miR-21 is associated with fibrosis and right ventricular failure. JCI Insight 2017, 2, e91625. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Charity, W.L.; Shi, W.; Gordon, K.S. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Kaisers, W.; Schwender, H.; Schaal, H. Hierarchical Clustering of DNA k-mer Counts in RNAseq Fastq Files Identifies Sample Heterogeneities. Int. J. Mol. Sci. 2018, 19, 3687. [Google Scholar] [CrossRef] [Green Version]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef] [Green Version]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Frigyesi, A.; Höglund, M. Non-negative matrix factorization for the analysis of complex gene expression data: Identification of clinically relevant tumor subtypes. Cancer Inform. 2008, 6, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Ahsan, M.; Mashuri, M.; Khusna, H.; Wibawati. Kernel principal component analysis (PCA) control chart for monitoring mixed non-linear variable and attribute quality characteristics. Heliyon 2022, 8, e09590. [Google Scholar] [CrossRef]
- Jiao, X.; Sherman, B.T.; Huang, W.D.; Robert, S.; Michael, W.B.; Clifford, H.L.; Richard, A.L. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Butler, H.; Cherr, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; Harris, M.A.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.; Wei, L.; et al. 0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Michael, K.; Stefan, W.; Milan, S.; Alberto, S.; Nadezhda, T.D.; Alexander, R.; Peer, B.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Rivera, C.G.; Vakil, R.; Bader, J.S. NeMo: Network Module identification in Cytoscape. BMC Bioinform. 2010, 11 (Suppl. 1), S61. [Google Scholar] [CrossRef] [Green Version]
- Doncheva, N.T.; Assenov, Y.; Domingues, F.S.; Albrecht, M. Topological analysis and interactive visualization of biological networks and protein structures. Nat. Protoc. 2012, 7, 670–685. [Google Scholar] [CrossRef]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Kang, L.; Wei, S.; Yang, C.; Fu, Y.; Ding, Z.; Zou, Y. Samm50 Promotes Hypertrophy by Regulating Pink1-Dependent Mitophagy Signaling in Neonatal Cardiomyocytes. Front. Cardiovasc. Med. 2021, 8, 748156. [Google Scholar] [CrossRef]
- Kim, S.C.; Stice, J.P.; Chen, L.; Jung, J.S.; Sanjiv, G.; Wang, Y.; Georg, B.; Joann, T.; Anne, A.K. Extracellular heat shock protein 60, cardiac myocytes, and apoptosis. Circ. Res. 2009, 105, 1186–1195. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; You, J.; Wang, X.; Wang, S.; Huang, J.; Xie, Q.; Gong, B.; Ding, Z.; Ye, Y.; Wang, C.; et al. Left ventricular response in the transition from hypertrophy to failure recapitulates distinct roles of Akt, β-arrestin-2, and CaMKII in mice with aortic regurgitation. Ann. Transl. Med. 2020, 8, 219. [Google Scholar] [CrossRef]
- Mukherjee, R.; Jow, L.; Noonan, D.; McDonnell, D.P. Human and rat peroxisome proliferator activated receptors (PPARs) demonstrate similar tissue distribution but different responsiveness to PPAR activators. J. Steroid Biochem. Mol. Biol. 1994, 51, 157–166. [Google Scholar] [CrossRef]
- Lee, S.S.; Pineau, T.; Drago, J.; Lee, E.J.; Owens, J.W.; Kroetz, D.L.; Fernandez-Salguero, P.M.; Westphal, H.; Gonzalez, F.J. Targeted disruption of the α isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol. Cell Biol. 1995, 15, 3012–3022. [Google Scholar] [CrossRef] [Green Version]
- Abbott, B.D. Review of the expression of peroxisome proliferator-activated receptors α (PPAR α), β (PPAR β), and γ (PPAR γ) in rodent and human development. Rep. Rod. Toxicol. 2009, 27, 246–257. [Google Scholar] [CrossRef]
- Li, S.; Yang, B.; Du, Y.; Lin, Y.; Liu, J.; Huang, S.; Zhang, A.; Jia, Z.; Zhang, Y. Targeting PPARα for the Treatment and Understanding of Cardiovascular Diseases. Cell Physiol. Biochem. 2018, 51, 2760–2775. [Google Scholar] [CrossRef]
- Newaz, M.; Blanton, A.; Fidelis, P.; Oyekan, A. NAD(P)H oxidase/nitric oxide interactions in peroxisome proliferator activated receptor (PPAR)α-mediated cardiovascular effects. Mutat. Res. 2005, 579, 163–171. [Google Scholar] [CrossRef]
- Ibarra-Lara, L.; Cervantes-Pérez, L.G.; Pérez-Severiano, F.; Valle, L.D.; Rubio-Ruíz, E.; Soria-Castro, E.; Pastelín-Hernández, G.S.; Sánchez-Aguilar, M.; Martínez-Lazcano, J.C.; Sánchez-Mendoza, A. PPARalpha stimulation exerts a blood pressure lowering effect through different mechanisms in a time-dependent manner. Eur. J. Pharmacol. 2010, 627, 185–193. [Google Scholar] [CrossRef]
- Touyz, R.M.; Schiffrin, E.L. Peroxisome proliferator-activated receptors in vascular biology-molecular mechanisms and clinical implications. Vascul. Pharmacol. 2006, 45, 19–28. [Google Scholar] [CrossRef]
- Brandt, J.M.; Djouadi, F.; Kelly, D.P. Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor α. J. Biol. Chem. 1998, 273, 23786–23792. [Google Scholar] [CrossRef] [Green Version]
- Remick, J.; Weintraub, H.; Setton, R.; Offenbacher, J.; Fisher, E.; Schwartzbard, A. Fibrate therapy: An update. Cardiol. Rev. 2008, 16, 129–141. [Google Scholar] [CrossRef]
- Gouni-Berthold, I.; Giannakidou, E.; Müller-Wieland, D.; Faust, M.; Kotzka, J.; Berthold, H.K.; Krone, W. Association between the PPARalpha L162V polymorphism, plasma lipoprotein levels, and atherosclerotic disease in patients with diabetes mellitus type 2 and in nondiabetic controls. Am. Heart J. 2004, 147, 1117–1124. [Google Scholar] [CrossRef]
- Arias, T.; Beaumont, J.; López, B.; Zalba, G.; Beloqui, O.; Barba, J.; Valencia, F.; Gómez-Doblas, J.J.; Teresa, E.D.; Díez, J. Association of the peroxisome proliferator-activated receptor α gene L162V polymorphism with stage C heart failure. J. Hypertens. 2011, 29, 876–883. [Google Scholar] [CrossRef]
- Gu, S.J.; Guo, Z.R.; Wu, M.; Ding, Y.; Luo, W.S. Association of peroxisome proliferator-activated receptor γ polymorphisms and haplotypes with essential hypertension. Genet. Test. Mol. Biomark. 2013, 17, 418–423. [Google Scholar] [CrossRef]
- Halder, I.; Champlin, J.; Sheu, L.; Goodpaster, B.H.; Manuck, S.B.; Ferrell, R.E.; Muldoon, M.F. PPARα gene polymorphisms modulate the association between physical activity and cardiometabolic risk. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 799–805. [Google Scholar] [CrossRef] [Green Version]
- Hirano, K.; Tanaka, T.; Ikeda, Y.; Yamaguchi, S.; Zaima, N.; Kobayashi, K.; Suzuki, A.; Sakata, Y.; Sakata, Y.; Kobayashi, K.; et al. Genetic mutations in adipose triglyceride lipase and myocardial up-regulation of peroxisome proliferated activated receptor-γ in patients with triglyceride deposit cardiomyovasculopathy. Biochem. Biophys. Res. Commun. 2014, 443, 574–579. [Google Scholar] [CrossRef]
- Muggenthaler, M.; Petropoulou, E.; Omer, S.; Simpson, M.A.; Sahak, H.; Rice, A.; Raju, H.; Conti, F.J.; Bridges, L.R.; Anderson, L.J.; et al. Whole exome sequence analysis reveals a homozygous mutation in PNPLA2 as the cause of severe dilated cardiomyopathy secondary to neutral lipid storage disease. Int. J. Cardiol. 2016, 210, 41–44. [Google Scholar] [CrossRef] [Green Version]
- Pasanisi, M.B.; Missaglia, S.; Cassandrini, D.; Salerno, F.; Farina, S.; Andreini, D.; Agostoni, P.; Morandi, L.; Mora, M.; Tavian, D. Severe cardiomyopathy in a young patient with complete deficiency of adipose triglyceride lipase due to a novel mutation in PNPLA2 gene. Int. J. Cardiol. 2016, 207, 165–167. [Google Scholar] [CrossRef] [Green Version]
- Rao, M.; Guo, G.; Li, M.; Chen, S.; Chen, K.; Chen, X.; Song, J.; Hu, S. The homozygous variant c. 245G > A/p. G82D in PNPLA2 is associated with arrhythmogenic cardiomyopathy phenotypic manifestations. Clin. Genet. 2019, 96, 532–540. [Google Scholar] [CrossRef]
- Alard, J.E.; Dueymes, M.; Mageed, R.A.; Saraux, A.; Youinou, P.; Jamin, C. Mitochondrial heat shock protein (HSP) 70 synergizes with HSP60 in transducing endothelial cell apoptosis induced by anti-HSP60 autoantibody. FASEB J. 2009, 23, 2772–2779. [Google Scholar] [CrossRef] [PubMed]
- Bodolay, E.; Prohászka, Z.; Paragh, G.; Csipő, I.; Nagy, G.; Laczik, R.; Demeter, N.; Zöld, E.; Nakken, B.; Szegedi, G.; et al. Increased levels of anti-heat-shock protein 60 (anti-Hsp60) indicate endothelial dysfunction, atherosclerosis and cardiovascular diseases in patients with mixed connective tissue disease. Immunol. Res. 2014, 60, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Rabczyński, M.; Fiodorenko-Dumas, Ż.; Mastej, K.; Dumas, I.; Adamiec, R.; Paprocka-Borowicz, M. A relationship between serological markers of chronic, C. pneumoniae and CMV infection and hsp60 in patients with atherosclerotic carotid stenosis. Acta Biochim. Pol. 2015, 62, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Wick, C. Tolerization against atherosclerosis using heat shock protein 60. Cell Stress Chaperones 2016, 21, 201–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentler, K.; Zhai, S.; Elsbecker, S.A.; Arnold, G.L.; Burton, B.K.; Vockley, J.; Cameron, C.A.; Hiner, S.J.; Edick, M.J.; Berry, S.A. 221 newborn-screened neonates with medium-chain acyl-coenzyme A dehydrogenase deficiency: Findings from the Inborn Errors of Metabolism Collaborative. Mol. Genet. Metab. 2016, 119, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonito, C.A.; Nunes, J.; Leandro, J.; Louro, F.; Leandro, P.; Ventura, F.V.; Guedes, R.C. Unveiling the Pathogenic Molecular Mechanisms of the Most Common Variant (p. K329E) in Medium-Chain Acyl-CoA Dehydrogenase Deficiency by in Vitro and in Silico Approaches. Biochemistry 2016, 55, 7086–7098. [Google Scholar] [CrossRef]
- Hara, K.; Tajima, G.; Okada, S.; Tsumura, M.; Kagawa, R.; Shirao, K.; Ohno, Y.; Yasunaga, S.; Ohtsubo, M.; Hata, I.; et al. Significance of ACADM mutations identified through newborn screening of MCAD deficiency in Japan. Mol. Genet. Metab. 2016, 118, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Davis, O.S.; Butcher, L.M.; Docherty, S.J.; Meaburn, E.L.; Curtis, C.J.; Simpson, M.A.; Schalkwyk, L.C.; Plomin, R. A three-stage genome-wide association study of general cognitive ability: Hunting the small effects. Behav. Genet. 2010, 40, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Hendrickson, S.L.; Lautenberger, J.A.; Chinn, L.W.; Malasky, M.; Sezgin, E.; Kingsley, L.A.; Goedert, J.J.; Kirk, G.D.; Gomperts, E.D.; Buchbinder, S.P.; et al. Genetic variants in nuclear-encoded mitochondrial genes influence AIDS progression. PLoS ONE 2010, 5, e12862. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Morinibu, A.; Kobayashi, M.; Zhu, Y.; Wang, X.; Goto, Y.; Yeom, C.J.; Zhao, T.; Hirota, K.; Shinomiya, K.; et al. Aberrant IDH3α expression promotes malignant tumor growth by inducing HIF-1-mediated metabolic reprogramming and angiogenesis. Oncogene 2015, 34, 4758–4766. [Google Scholar] [CrossRef] [Green Version]
- May, J.L.; Kouri, F.M.; Hurley, L.A.; Liu, J.; Tommasini-Ghelfi, S.; Ji, Y.; Gao, P.; Calvert, A.E.; Lee, A.; Chandel, N.S.; et al. IDH3α regulates one-carbon metabolism in glioblastoma. Sci. Adv. 2019, 5, eaat0456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, V.G.; Nikopoulos, K.; Quinodoz, M.; Granse, L.; Farinelli, P.; Superti-Furga, A.; Andréasson, S.; Rivolta, C. A novel missense variant in IDH3A causes autosomal recessive retinitis pigmentosa. Ophthalmic Genet. 2019, 40, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, W.; Leung, P.S.; Bowlus, C.L.; Dhaliwal, S.; Coppel, R.L.; Ansari, A.A.; Yang, G.X.; Wang, J.; Kenny, T.P.; et al. Ongoing activation of autoantigen-specific B cells in primary biliary cirrhosis. Hepatology 2014, 60, 1708–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellucci, R.; Oertelt, S.; Gallagher, M.; Li, S.; Zorn, E.; Weller, E.; Porcheray, F.; Alyea, E.P.; Soiffer, R.J.; Munshi, N.C.; et al. Differential epitope mapping of antibodies to PDC-E2 in patients with hematologic malignancies after allogeneic hematopoietic stem cell transplantation and primary biliary cirrhosis. Blood 2007, 109, 2001–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, C.; Elf, S.; Ji, Q.; Kang, H.B.; Zhou, L.; Hitosugi, T.; Jin, L.; Lin, R.; Zhang, L.; Seo, J.H.; et al. Lysine acetylation activates 6-phosphogluconate dehydrogenase to promote tumor growth. Mol. Cell 2014, 55, 552–565. [Google Scholar] [CrossRef] [Green Version]
- Stegbauer, J.; Kraus, M.; Nordmeyer, S.; Kirchner, M.; Ziehm, M.; Dommisch, H.; Kelle, S.; Kelm, M.; Baczko, I.; Landmesser, U.; et al. Proteomic Analysis Reveals Upregulation of ACE2 (Angiotensin-Converting Enzyme 2), the Putative SARS-CoV-2 Receptor in Pressure-but Not Volume-Overloaded Human Hearts. Hypertension 2020, 76, e41–e43. [Google Scholar] [CrossRef]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef]
- Rong, J.; Tao, X.; Lin, Y.; Zheng, H.; Ning, L.; Lu, H.S.; Daugherty, A.; Shi, P.; Mullick, A.E.; Chen, S.; et al. Loss of Hepatic Angiotensinogen Attenuates Sepsis-Induced Myocardial Dysfunction. Circ. Res. 2021, 129, 547–564. [Google Scholar] [CrossRef]
- Sanchis-Gomar, F.; Lavie, C.J.; Perez-Quilis, C.; Henry, B.M.; Lippi, G. Angiotensin-Converting Enzyme 2 and Antihypertensives (Angiotensin Receptor Blockers and Angiotensin-Converting Enzyme Inhibitors) in Coronavirus Disease 2019. Mayo Clin. Proc. 2020, 95, 1222–1230. [Google Scholar] [CrossRef]
- Patel, V.B.; Clarke, N.; Wang, Z.; Fan, D.; Parajuli, N.; Basu, R.; Putko, B.; Kassiri, Z.; Turner, A.J.; Oudit, G.Y. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: A positive feedback mechanism in the RAS. J. Mol. Cell Cardiol. 2014, 66, 167–176. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Gemmati, D.; Tisato, V. Genetic Hypothesis and Pharmacogenetics Side of Renin-Angiotensin-System in COVID-19. Genes 2020, 11, 1044. [Google Scholar] [CrossRef] [PubMed]
- Milani, D.; Caruso, L.; Zauli, E.; Al Owaifeer, A.M.; Secchiero, P.; Zauli, G.; Gemmati, D.; Tisato, V. p53/NF-kB Balance in SARS-CoV-2 Infection: From OMICs, Genomics and Pharmacogenomics Insights to Tailored Therapeutic Perspectives (COVIDomics). Front. Pharmacol. 2022, 13, 871583. [Google Scholar] [CrossRef]
- Zhao, M.; Liu, Z.; Shao, F.; Zhou, W.; Chen, Z.; Xia, P.; Wang, S.; Yang, P. Communication Pattern Changes Along With Declined IGF1 of Immune Cells in COVID-19 Patients During Disease Progression. Front. Immunol. 2021, 12, 729990. [Google Scholar] [CrossRef] [PubMed]
- Pfeiler, G.; DeMichele, A.; Dueck, A.C.; Fesl, C.; Gnant, M.; Mayer, E.L. Safety of adjuvant CDK4/6 inhibitors during the COVID-19 pandemic. Lancet Oncol. 2022, 23, 195–197. [Google Scholar] [CrossRef]
- Leri, A.; Liu, Y.; Claudio, P.P.; Kajstura, J.; Wang, X.; Wang, S.; Kang, P.; Malhotra, A.; Anversa, P. Insulin-like growth factor-1 induces Mdm2 and down-regulates p53, attenuating the myocyte renin-angiotensin system and stretch-mediated apoptosis. Am. J. Pathol. 1999, 154, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Leri, A.; Liu, Y.; Wang, X.; Kajstura, J.; Malhotra, A.; Meggs, L.G.; Anversa, P. Overexpression of insulin-like growth factor-1 attenuates the myocyte renin-angiotensin system in transgenic mice. Circ. Res. 1999, 84, 752–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- South, A.M.; Diz, D.I.; Chappell, M.C. COVID-19, ACE2, and the cardiovascular consequences. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1084–H1090. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GSE Series | Type | Sample Size | Platform | ||
|---|---|---|---|---|---|
| Control | Pulmonary Insufficiency and Stenosis | Transverse Aortic Constriction | |||
| GSE97363 | mRNA | 10 | 4 | GPL13912 | |
| GSE52796 | mRNA | 3 | 5 | GPL6887 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, Y.; Zhao, D.; Zhou, Y.; Lu, J.; Kang, L.; Jiang, X.; Xu, R.; Ding, Z.; Zou, Y. Identification of Differential Expression Genes between Volume and Pressure Overloaded Hearts Based on Bioinformatics Analysis. Genes 2022, 13, 1276. https://doi.org/10.3390/genes13071276
Fu Y, Zhao D, Zhou Y, Lu J, Kang L, Jiang X, Xu R, Ding Z, Zou Y. Identification of Differential Expression Genes between Volume and Pressure Overloaded Hearts Based on Bioinformatics Analysis. Genes. 2022; 13(7):1276. https://doi.org/10.3390/genes13071276
Chicago/Turabian StyleFu, Yuanfeng, Di Zhao, Yufei Zhou, Jing Lu, Le Kang, Xueli Jiang, Ran Xu, Zhiwen Ding, and Yunzeng Zou. 2022. "Identification of Differential Expression Genes between Volume and Pressure Overloaded Hearts Based on Bioinformatics Analysis" Genes 13, no. 7: 1276. https://doi.org/10.3390/genes13071276
APA StyleFu, Y., Zhao, D., Zhou, Y., Lu, J., Kang, L., Jiang, X., Xu, R., Ding, Z., & Zou, Y. (2022). Identification of Differential Expression Genes between Volume and Pressure Overloaded Hearts Based on Bioinformatics Analysis. Genes, 13(7), 1276. https://doi.org/10.3390/genes13071276