
Chromosomal Translocations Detection in Cancer Cells Using Chromosomal Conformation Capture Data

 ,
, 
Abstract
:
1. Introduction
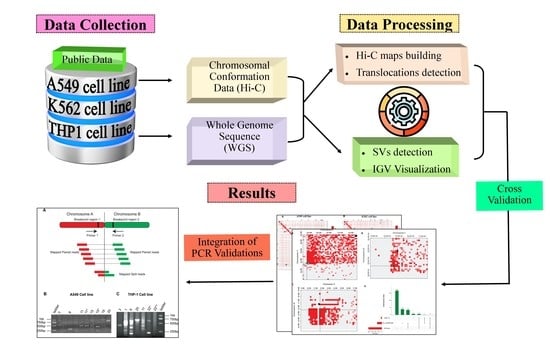
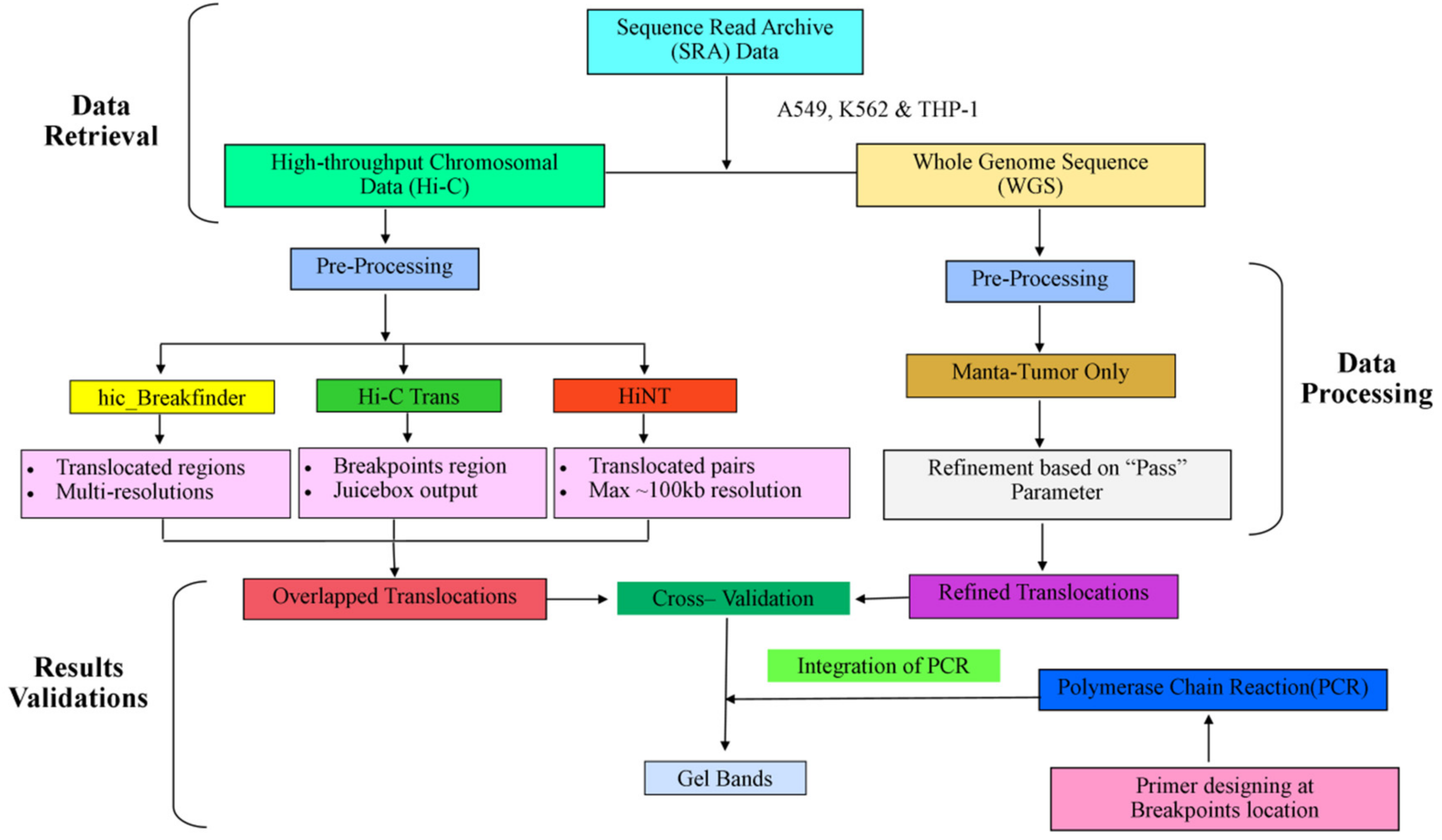
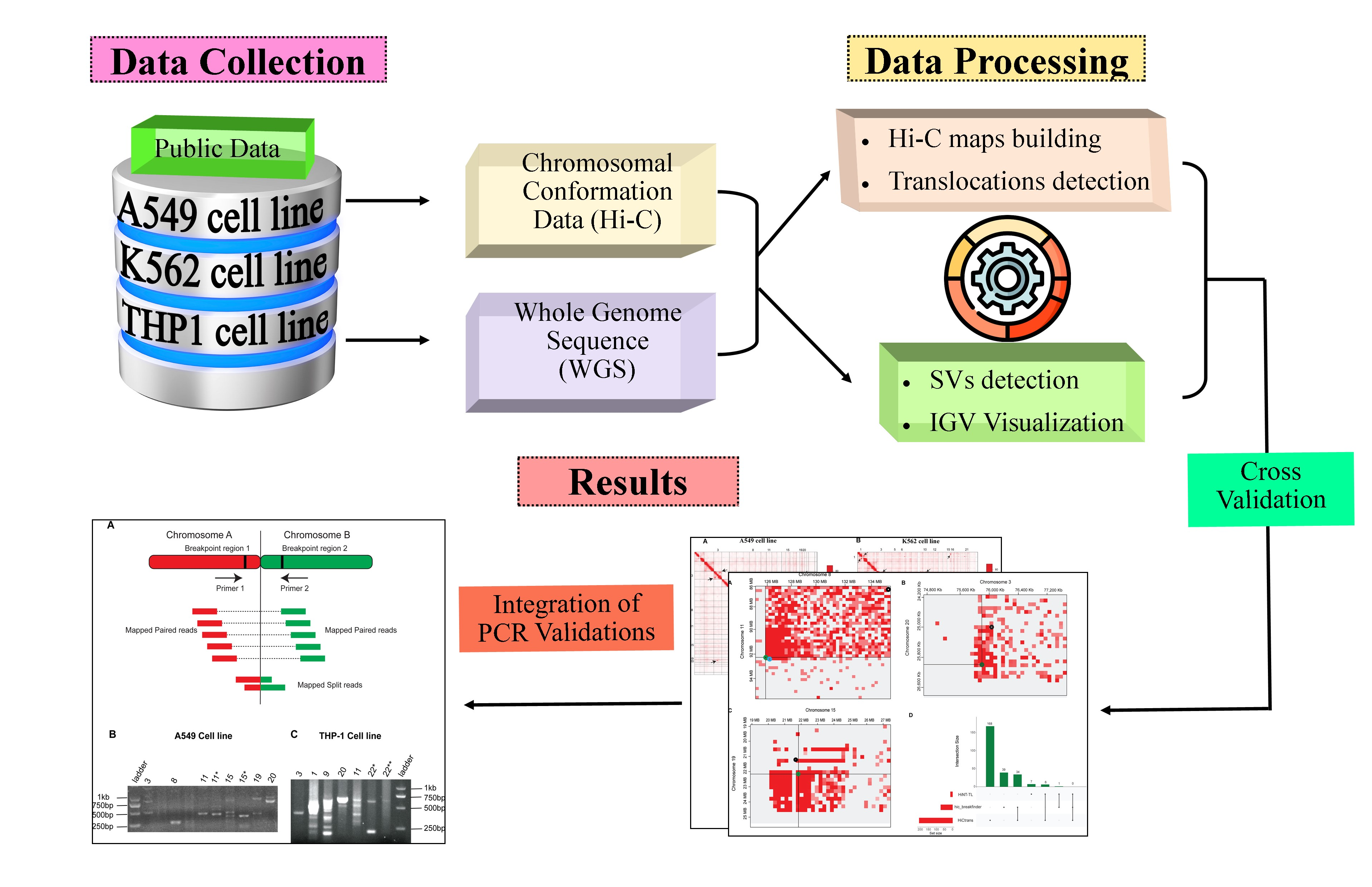
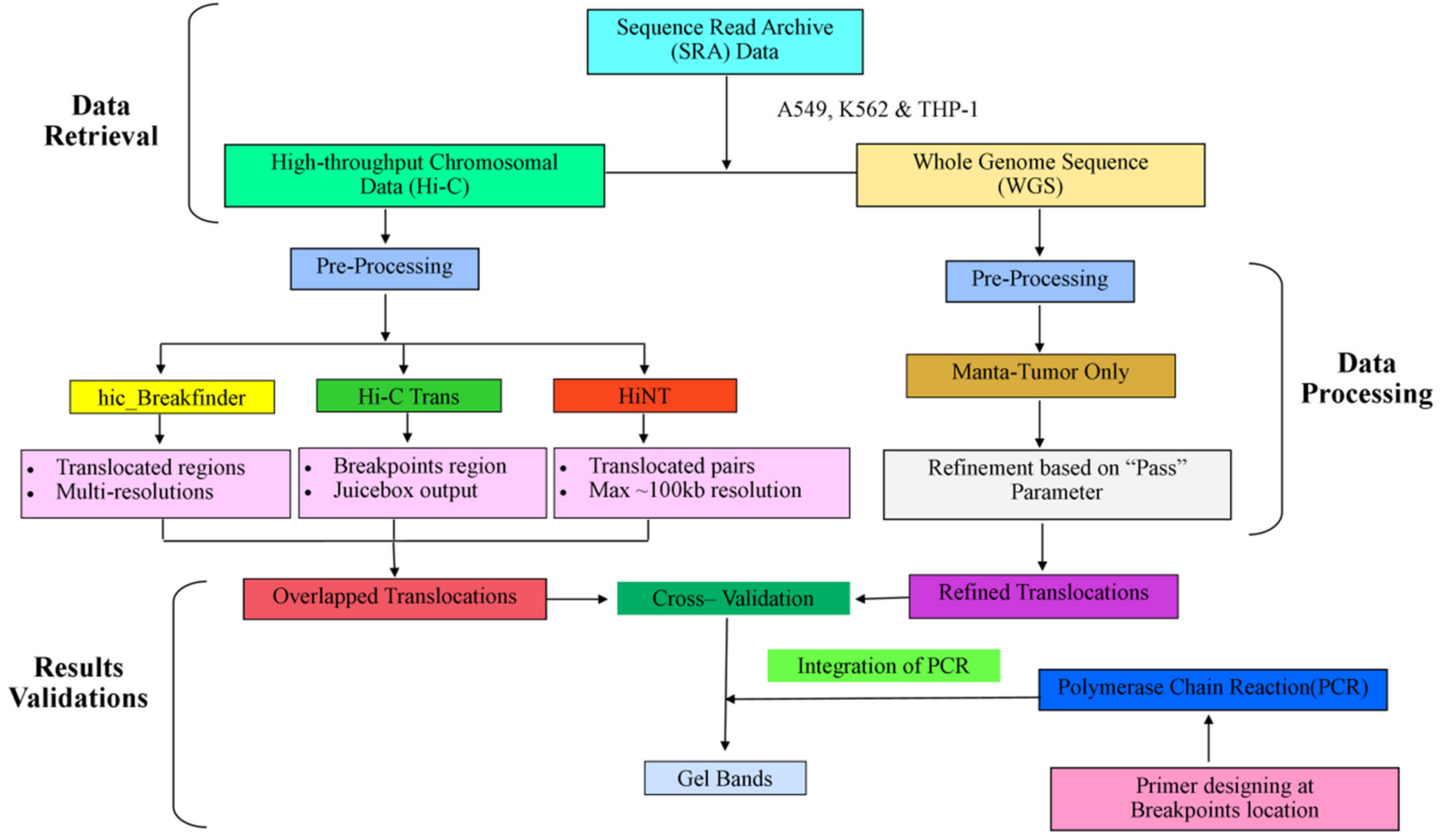
2. Materials and Methods
2.1. Data Source
2.2. Hi-C Data Processing
2.3. Translocations Detection
2.4. Whole-Genome Sequence Processing
2.5. PCR Validation
3. Results
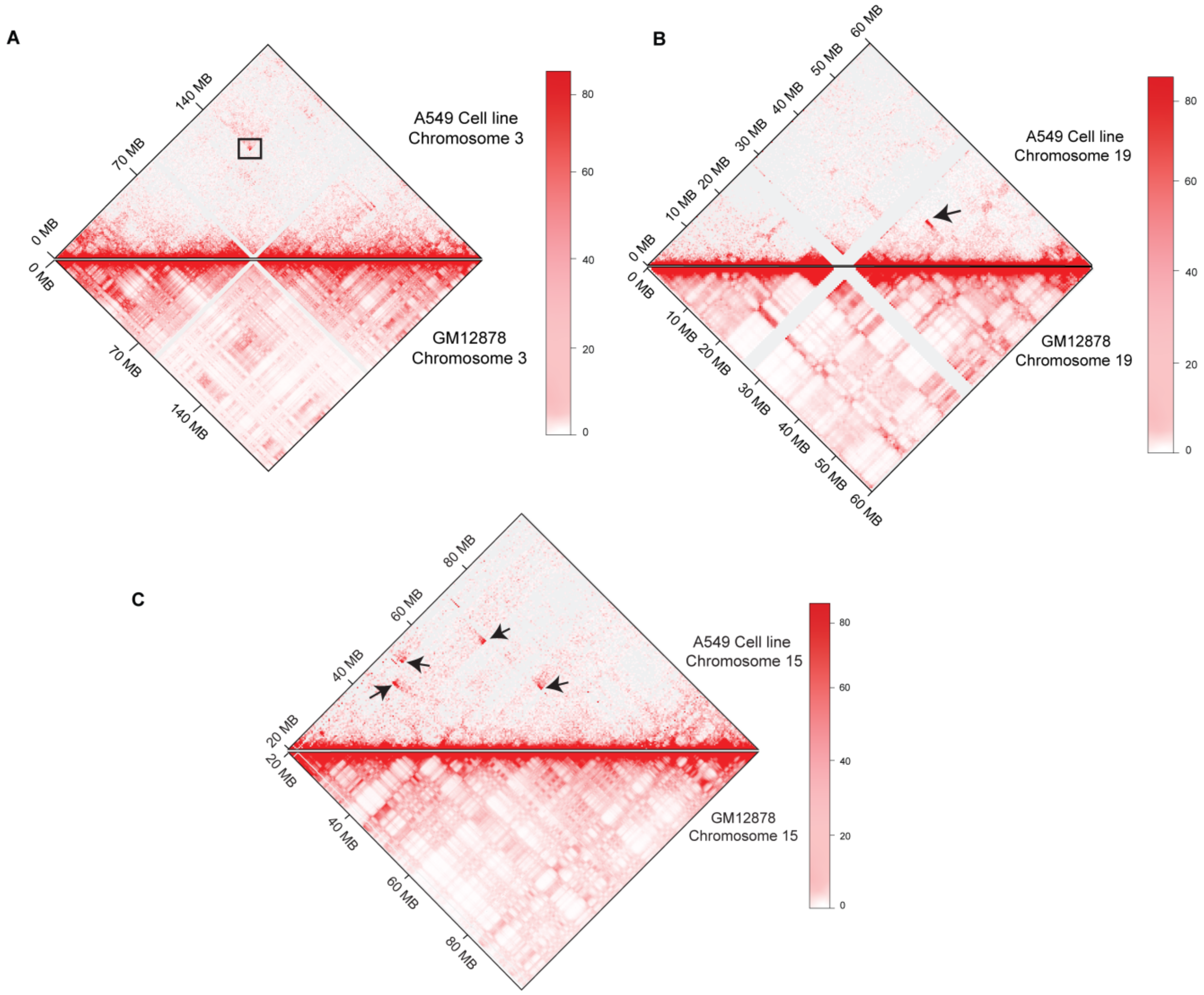
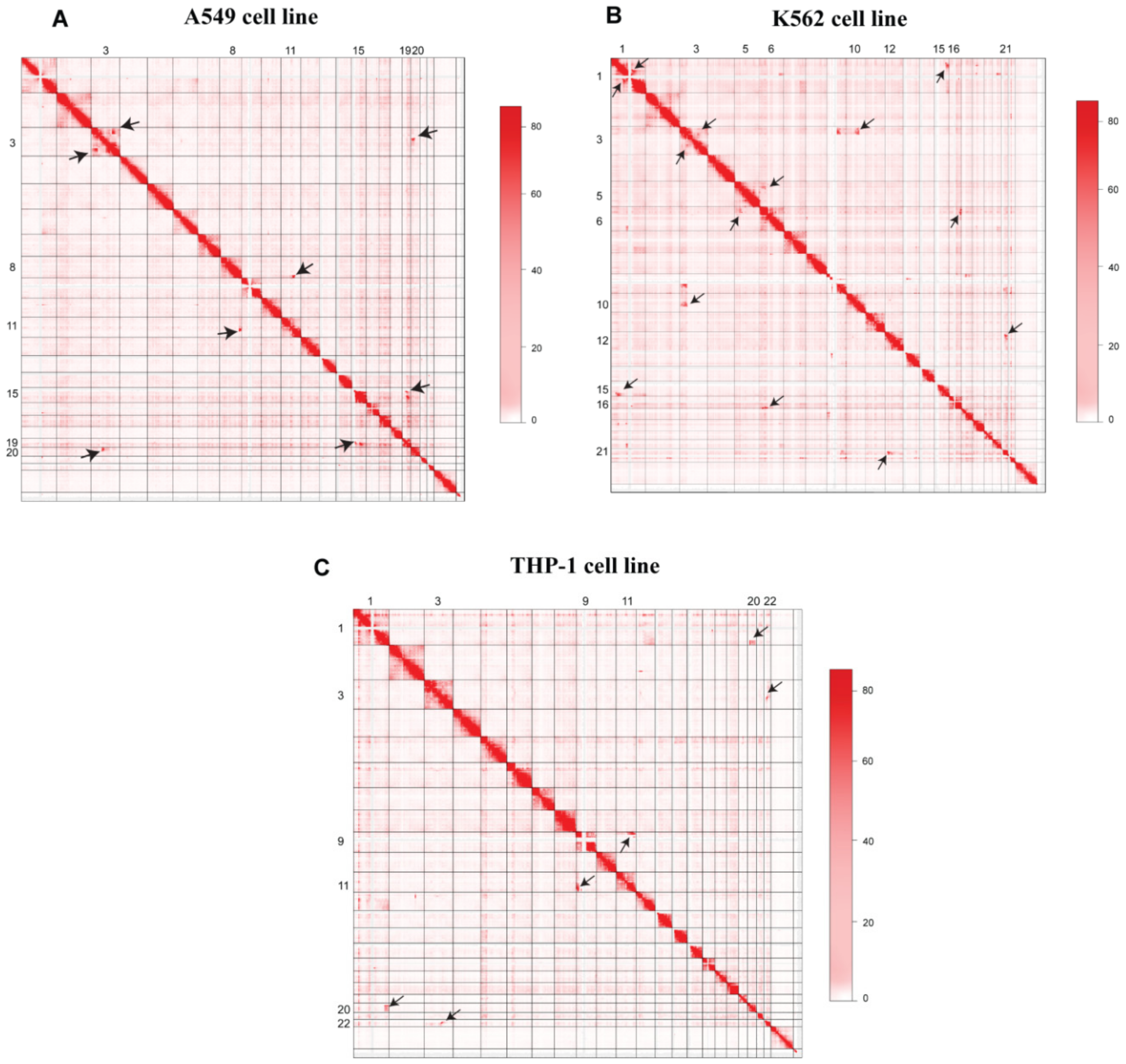
3.1. 3D-Genome Variations across Different Cell Lines
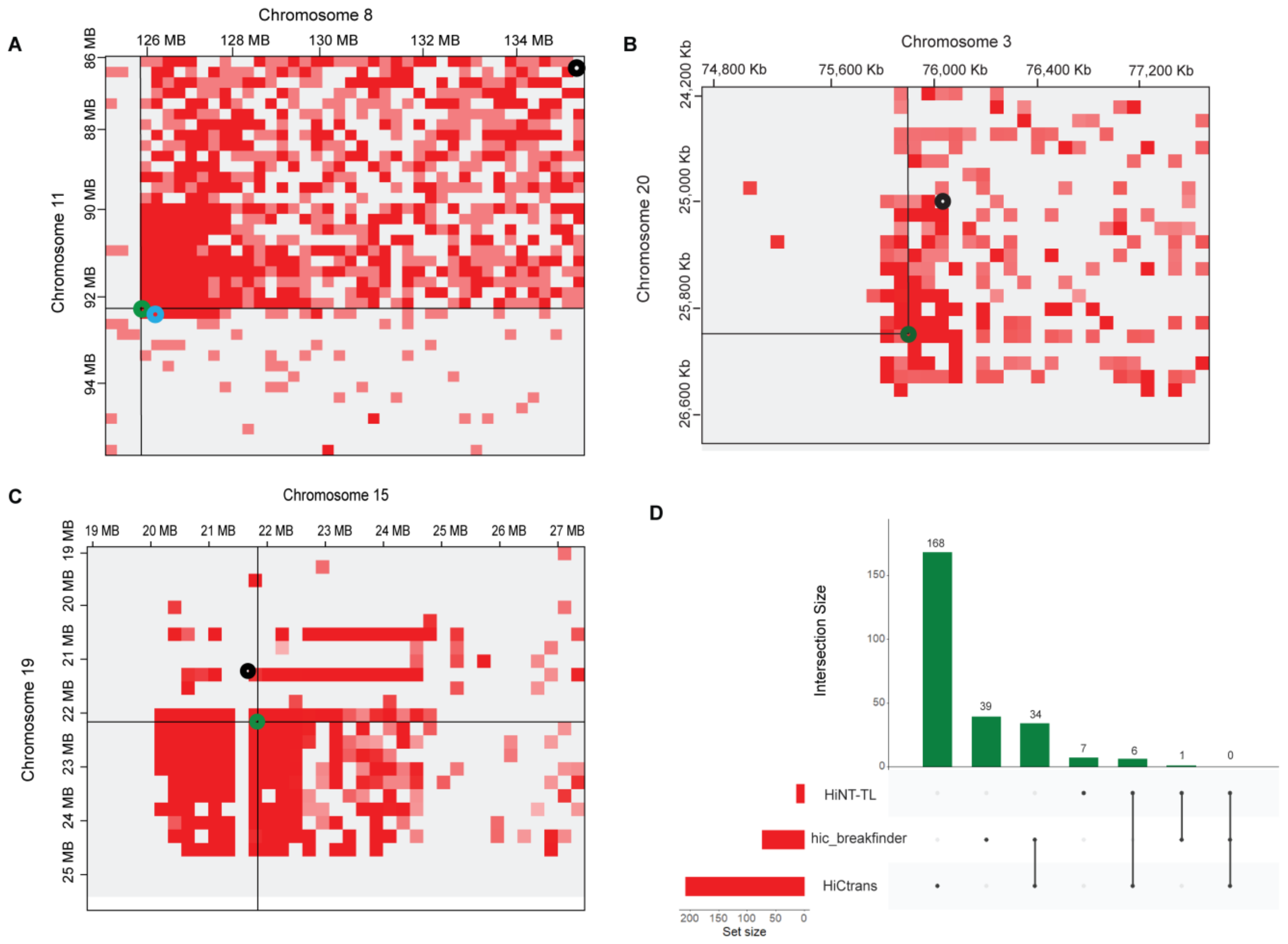
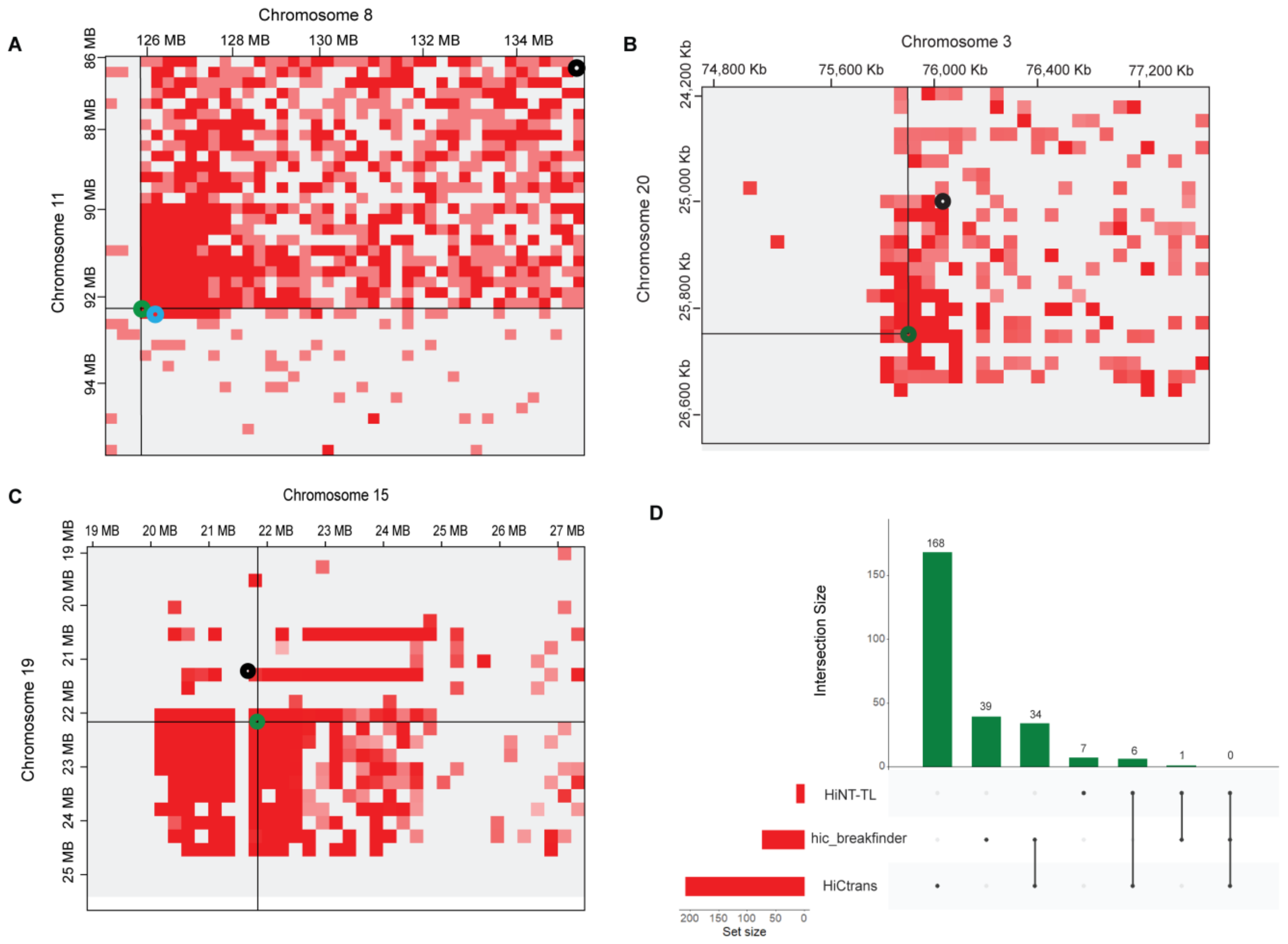
3.2. High-Resolution Translocations Detection
3.3. Cross-Validation of Translocations in WGS Data
3.4. PCR Amplification
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Sadowski, M.; Kraft, A.; Szalaj, P.; Wlasnowolski, M.; Tang, Z.; Ruan, Y.; Plewczynski, D. Spatial chromatin architecture alteration by structural variations in human genomes at the population scale. Genome Biol. 2019, 20, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Fantes, J.A.; Boland, E.; Ramsay, J.; Donnai, D.; Splitt, M.; Goodship, J.A.; Stewart, H.; Whiteford, M.; Gautier, P.; Harewood, L.; et al. FISH Mapping of De Novo Apparently Balanced Chromosome Rearrangements Identifies Characteristics Associated with Phenotypic Abnormality. Am. J. Hum. Genet. 2008, 82, 916–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.Y.; et al. An integrated map of structural variation in 2,504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Soemedi, R.; Wilson, I.J.; Bentham, J.; Darlay, R.; Töpf, A.; Zelenika, D.; Cosgrove, C.; Setchfield, K.; Thornborough, C.; Granados-Riveron, J.; et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am. J. Hum. Genet. 2012, 91, 489–501. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Roos, J.L.; Levy, S.; Van Rensburg, E.J.; Gogos, J.A.; Karayiorgou, M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 2008, 40, 880–885. [Google Scholar] [CrossRef]
- Spielmann, M.; Lupiáñez, D.G.; Mundlos, S. Structural variation in the 3D genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef] [Green Version]
- Belkadi, A.; Bolze, A.; Itan, Y.; Cobat, A.; Vincent, Q.B.; Antipenko, A.; Shang, L.; Boisson, B.; Casanova, J.L.; Abel, L. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 5473–5478. [Google Scholar] [CrossRef] [Green Version]
- Sud, A.; Kinnersley, B.; Houlston, R.S. Genome-wide association studies of cancer: Current insights and future perspectives. Nat. Rev. Cancer 2017, 17, 692–704. [Google Scholar] [CrossRef]
- Chaisson, M.J.P.; Huddleston, J.; Dennis, M.Y.; Sudmant, P.H.; Malig, M.; Hormozdiari, F.; Antonacci, F.; Surti, U.; Sandstrom, R.; Boitano, M.; et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 2015, 517, 608–611. [Google Scholar] [CrossRef] [Green Version]
- Pendleton, M.; Sebra, R.; Pang, A.W.C.; Ummat, A.; Franzen, O.; Rausch, T.; Stütz, A.M.; Stedman, W.; Anantharaman, T.; Hastie, A.; et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat. Methods 2015, 12, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.S.; Rhie, A.; Kim, J.; Lee, S.; Sohn, M.H.; Kim, C.U.; Hastie, A.; Cao, H.; Yun, J.Y.; Kim, J.; et al. De novo assembly and phasing of a Korean human genome. Nature 2016, 538, 243–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- English, A.C.; Salerno, W.J.; Reid, J.G. PBHoney: Identifying genomic variants via long-read discordance and interrupted mapping. BMC Bioinform. 2014, 15, 180. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.; Ay, F. Identification of copy number variations and translocations in cancer cells from Hi-C data. Bioinformatics 2018, 34, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Adeel, M.M.; Jiang, H.; Arega, Y.; Cao, K.; Lin, D.; Cao, C.; Cao, G.; Wu, P.; Li, G. Structural Variations of the 3D Genome Architecture in Cervical Cancer Development. Front. Cell Dev. Biol. 2021, 9, 1885. [Google Scholar] [CrossRef]
- Lin, D.; Hong, P.; Zhang, S.; Xu, W.; Jamal, M.; Yan, K.; Lei, Y.; Li, L.; Ruan, Y.; Fu, Z.F.; et al. Digestion-ligation-only Hi-C is an efficient and cost-effective method for chromosome conformation capture. Nat. Genet. 2018, 50, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 February 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Hong, P.; Jiang, H.; Xu, W.; Lin, D.; Xu, Q.; Cao, G.; Li, G. DLO Hi-C Tool for Digestion-Ligation-Only Hi-C Chromosome Conformation Capture Data Analysis. Genes 2019, 11, 289. [Google Scholar] [CrossRef] [Green Version]
- Servant, N.; Varoquaux, N.; Lajoie, B.R.; Viara, E.; Chen, C.J.; Vert, J.P.; Heard, E.; Dekker, J.; Barillot, E. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015, 16, 259. [Google Scholar] [CrossRef] [Green Version]
- Imakaev, M.; Fundenberg, G.; McCord, R.P.; Naumova, N.; Goloborodko, N.; Lajoie, B.R.; Dekker, J.; Mirny, L.A. Iterative Correction of Hi-C Data Reveals Hallmarks of Chromosome Organization. Nat. Methods 2012, 9, 999–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speicher, M.R.; Ballard, S.G.; Ward, D.C. Karyotyping human chromosomes by combinatorial multi-fluor FISH. Nat. Genet. 1996, 12, 368–375. [Google Scholar] [CrossRef] [PubMed]
- LaFramboise, T. Single nucleotide polymorphism arrays: A decade of biological, computational and technological advances. Nucleic Acids Res. 2009, 37, 4181–4193. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Lee, S.; Chu, C.; Jain, D.; Kerpedjiev, P.; Nelson, G.M.; Walsh, J.M.; Alver, B.H.; Park, P.J. HiNT: A computational method for detecting copy number variations and translocations from Hi-C data. Genome Biol. 2020, 21, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durand, N.C.; Robinson, J.T.; Shamim, M.S.; Machol, I.; Mesirov, J.P.; Lander, E.S.; Aiden, E.L. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 2016, 3, 99–101. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genome Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Dixon, J.R.; Xu, J.; Dileep, V.; Zhan, Y.; Song, F.; Le, V.T.; Yardımcı, G.G.; Chakraborty, A.; Bann, D.V.; Wang, Y.; et al. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet. 2018, 50, 1388–1398. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Liang, F.; Cheng, D.; Zhang, Z.; Yu, G.; Zha, J.; Wang, Y.; Xia, Q.; Yuan, D.; Tan, Y.; et al. Location of Balanced Chromosome-Translocation Breakpoints by Long-Read Sequencing on the Oxford Nanopore Platform. Front. Genet. 2020, 10, 1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Vissers, L.E.L.M.; Van Ravenswaaij, C.M.A.; Admiraal, R.; Hurst, J.A.; De Vries, B.B.A.; Janssen, I.M.; Van Der Vliet, W.A.; Huys, E.H.L.P.G.; De Jong, P.J.; Hamel, B.C.J.; et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 2004, 36, 955–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lelieveld, S.H.; Spielmann, M.; Mundlos, S.; Veltman, J.A.; Gilissen, C. Comparison of Exome and Genome Sequencing Technologies for the Complete Capture of Protein-Coding Regions. Hum. Mutat. 2015, 36, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Hayes, M.; Li, J. Bellerophon: A hybrid method for detecting interchromo-somal rearrangements at base pair resolution using next-generation sequencing data. BMC Bioinform. 2013, 14, S6. [Google Scholar] [CrossRef] [Green Version]
- Harewood, L.; Kishore, K.; Eldridge, M.D.; Wingett, S.; Pearson, D.; Schoenfelder, S.; Collins, V.P.; Fraser, P. Hi-C as a tool for precise detection and characterisation of chromosomal rearrangements and copy number variation in human tumours. Genome Biol. 2017, 18, 125. [Google Scholar] [CrossRef] [Green Version]
- Dang, T.P.; Gazdar, A.F.; Virmani, A.K.; Sepetavec, T.; Hande, K.R.; Minna, J.D.; Roberts, J.R.; Carbone, D.P. Chromosome 19 translocation, overexpression of Notch3, and human lung cancer. J. Natl. Cancer Inst. 2000, 92, 1355–1357. [Google Scholar] [CrossRef] [Green Version]
- Rohrbeck, A.; Borlak, J. Cancer genomics identifies regulatory gene networks associated with the transition from dysplasia to advanced lung adenocarcinomas induced by c-Raf-1. PLoS ONE 2009, 4, e7315. [Google Scholar] [CrossRef]
- Li, R.; Yang, Y.E.; Yin, Y.H.; Zhang, M.Y.; Li, H.; Qu, Y.Q. Methylation and transcriptome analysis reveal lung adenocarcinoma-specific diagnostic biomarkers. J. Transl. Med. 2019, 17, 324. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhang, Y.H.; Zhang, Z.; Huang, T.; Cai, Y.D. Inferring Novel Tumor Suppressor Genes with a Protein-Protein Interaction Network and Network Diffusion Algorithms. Mol. Ther. Methods Clin. Dev. 2018, 10, 57–67. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Agarwala, V.; Mirny, L.A. Three-Dimensional Genome Architecture Influences Partner Selection for Chromosomal Translocations in Human Disease. PLoS ONE 2012, 7, e44196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, E.C.; Grand, R.S.; Perry, J.K.; Vickers, M.H.; Olins, A.L.; Olins, D.E.; O’Sullivan, J.M. Hi-C detects novel structural variants in HL-60 and HL-60/S4 cell lines. Genomics 2020, 112, 151–162. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, G.A.; Kats, L.; Pandolfi, P.P. Synergy against PML-RARa: Targeting transcription, proteolysis, differentiation, and self-renewal in acute promyelocytic leukemia. J. Exp. Med. 2013, 210, 2793–2802. [Google Scholar] [CrossRef] [PubMed]
- Sattler, M.; Griffin, J.D. Mechanisms of transformation by the BCR/ABL oncogene. Int. J. Hematol. 2001, 73, 278–291. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Exp.ID/Accession | Data Type | Data Availability |
|---|---|---|---|
| A549 | Lab data | In situ Hi-C_rep1 | In-house |
| A549 | Lab data | In situ Hi-C_rep2 | In-house |
| A549 | SRR14843382 | WGS | Public |
| K562 | SRR6468005 | DLO Hi-C_rep1 | Public |
| K562 | SRR6468006 | DLO Hi-C_rep2 | Public |
| K562 | SRR14828339 | WGS | Public |
| THP-1 | SRR5005053 | DLO Hi-C | Public |
| THP-1 | SRR8670675 | WGS | Public |
| Cell Line | Hi-C Breakpoints | WGS Breakpoints | PCR |
|---|---|---|---|
| A549 | Chr11:92110000-92340000 Chr8:125920000-125950000 | Chr11:92232864-92337991 Chr8:125915486-125920628 | √ |
| A549 | Chr3:69600000-76000000 Chr20:24900000-26500000 | Chr3:759889069-75989055 Chr20:26204286-26204887 | √ |
| A549 | Chr15:22298000-22368000 Chr19:22005000-22019000 | Chr15:22290005-22291154 Chr19:22004010-22005005 | √ |
| K562 | Chr1:34540000-34750000 Chr15:79000000-79220000 | Chr1:164354987-164355279 Chr15:79163087-79163379 | × |
| K562 | Chr3:48257000-48287000 Chr10:87878000-87989000 | Chr3:48227444-48228179 Chr10:87848597-87849236 | √ |
| K562 | Chr5:21560000-21575000 Chr6:57680000-57760000 | Chr5:21573105-21573417 Chr6:57575670-57575919 | √ |
| K562 | Chr12:22560000-22566000 Chr21:25532000-25534001 | Chr12:22711223-22711519 Chr21:25504958-25505270 | √ |
| K562 | Chr6:16753000-16768000 Chr16:85549000-85567000 | Chr6:33618899-33619146 Chr16:63908450-63908595 | × |
| THP-1 | Chr9:14520000-15460000 Chr11:118350000-118900000 | Chr9:15458220-15458580 Chr11:118356100-118356400 | √ |
| THP-1 | Chr22: 29020000-29060000 Chr3: 119580000-119670000 | Chr22:29081888-29081988 Chr3:119651343-119651367 | √ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adeel, M.M.; Rehman, K.; Zhang, Y.; Arega, Y.; Li, G. Chromosomal Translocations Detection in Cancer Cells Using Chromosomal Conformation Capture Data. Genes 2022, 13, 1170. https://doi.org/10.3390/genes13071170
Adeel MM, Rehman K, Zhang Y, Arega Y, Li G. Chromosomal Translocations Detection in Cancer Cells Using Chromosomal Conformation Capture Data. Genes. 2022; 13(7):1170. https://doi.org/10.3390/genes13071170
Chicago/Turabian StyleAdeel, Muhammad Muzammal, Khaista Rehman, Yan Zhang, Yibeltal Arega, and Guoliang Li. 2022. "Chromosomal Translocations Detection in Cancer Cells Using Chromosomal Conformation Capture Data" Genes 13, no. 7: 1170. https://doi.org/10.3390/genes13071170
APA StyleAdeel, M. M., Rehman, K., Zhang, Y., Arega, Y., & Li, G. (2022). Chromosomal Translocations Detection in Cancer Cells Using Chromosomal Conformation Capture Data. Genes, 13(7), 1170. https://doi.org/10.3390/genes13071170