
Identification and Functional Analysis of Individual-Specific Subpathways in Lung Adenocarcinoma

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Preparation
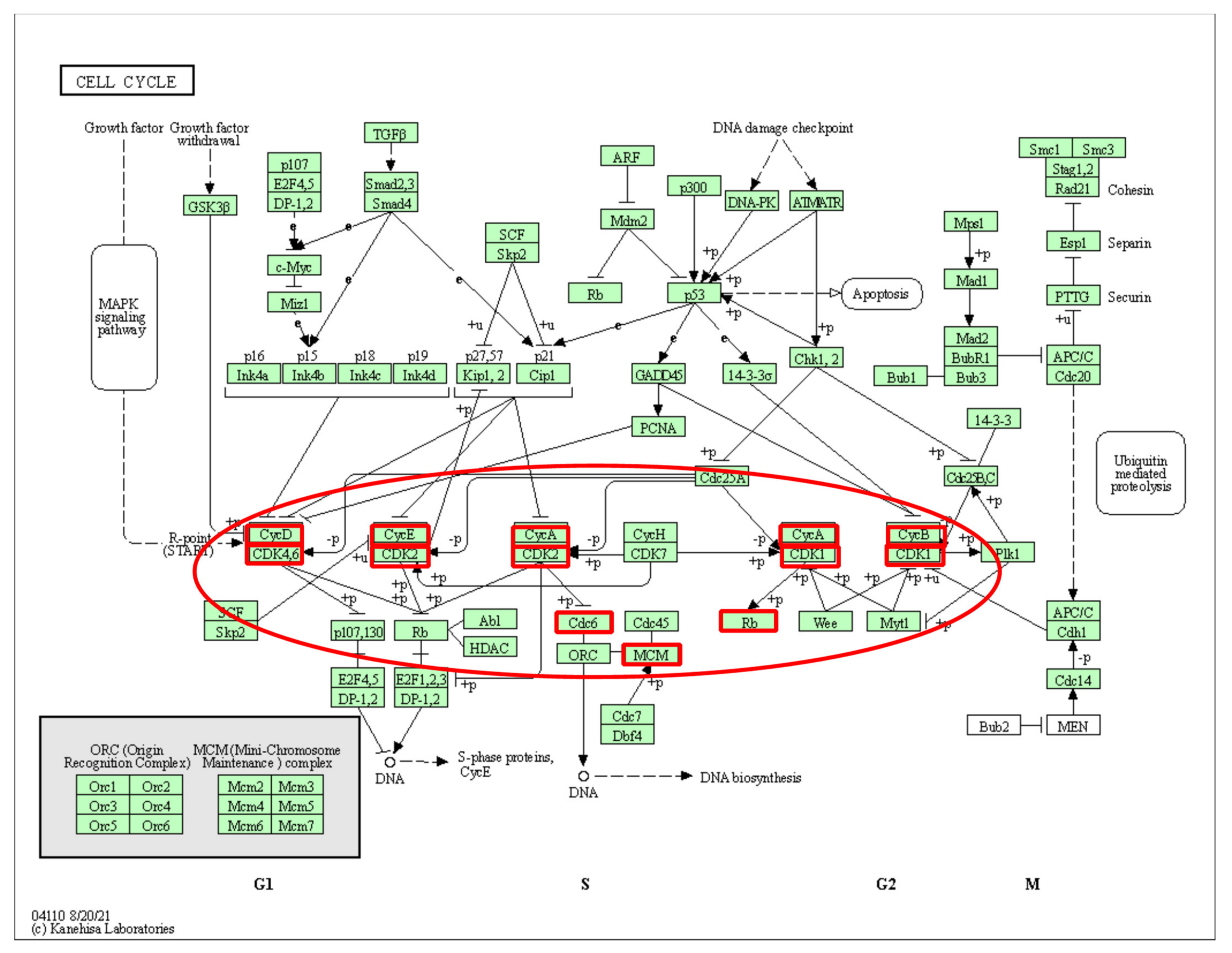
2.2. Pathway Information
2.3. Overview of the Subpathway Identification Pipeline
2.4. Calculation of Edge Scores
2.5. Subpathway Extraction
2.6. Survival Analysis
3. Results
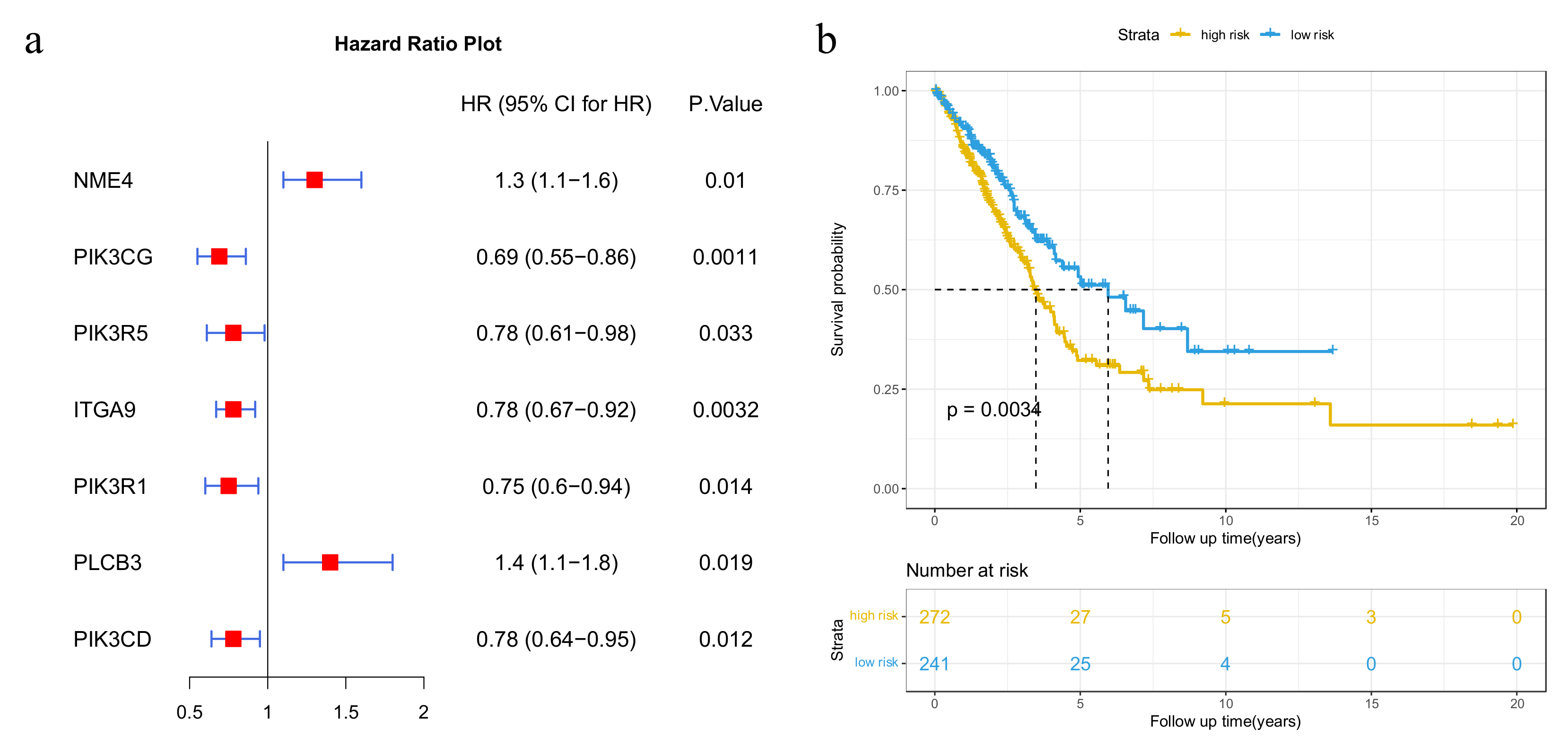
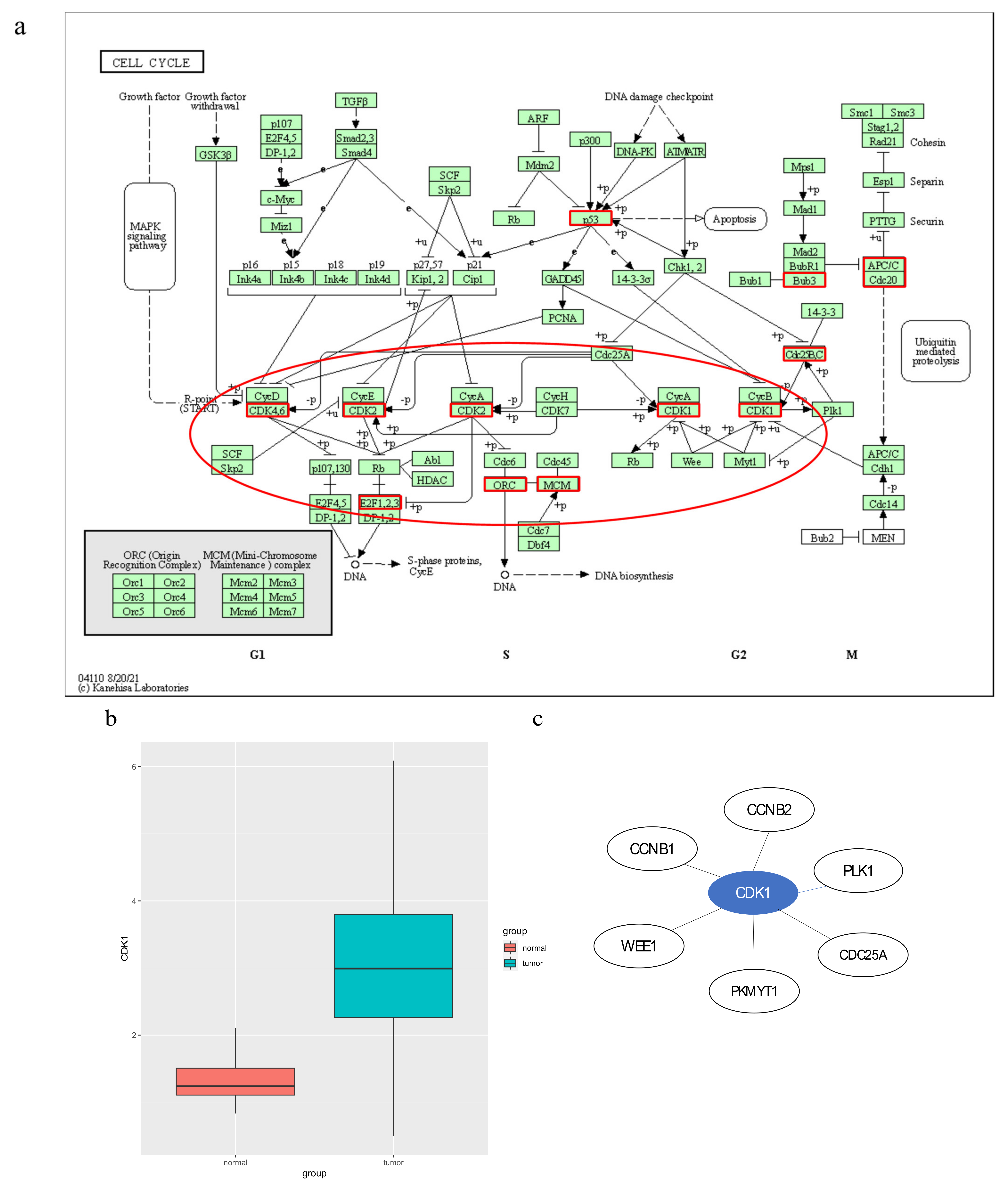
3.1. Mutated Genes in Subpathways
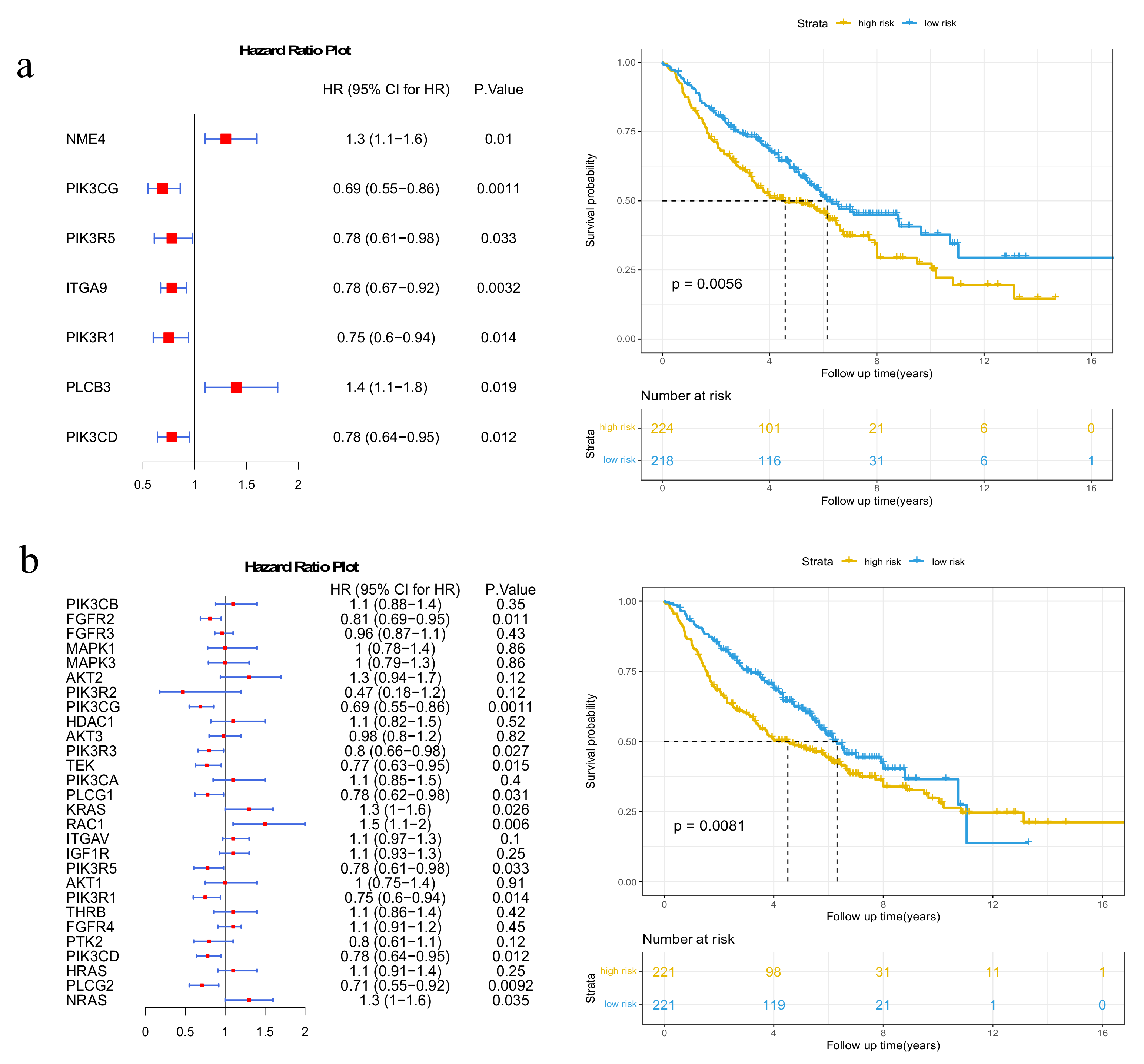
3.2. Potential Disease Genes in Subpathways
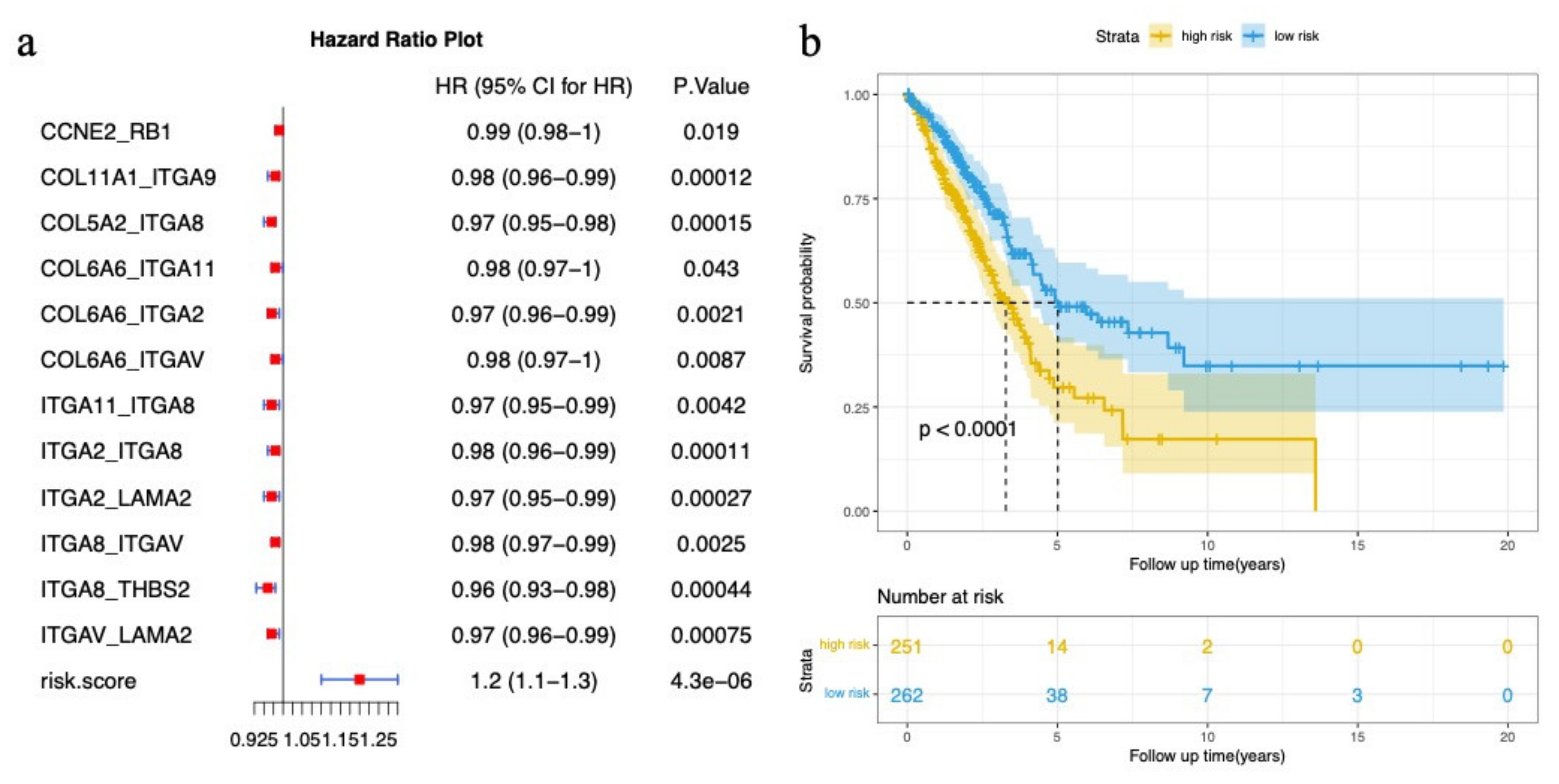
3.3. Immune-Related Genes in Subpathways
3.4. Cancer-Related Subpathway Identification
3.5. Validation on GEO Dataset
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Tian, W.J.; Liu, S.S.; Li, B.R. The Combined Detection of Immune Genes for Predicting the Prognosis of Patients With Non-Small Cell Lung Cancer. Technol. Cancer Res. Treat. 2020, 19, 1533033820977504. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, J.; Zhan, X. Identification of Immune-Related Gene Signatures in Lung Adenocarcinoma and Lung Squamous Cell Carcinoma. Front. Immunol. 2021, 12, 752643. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yang, Y.E.; Yin, Y.H.; Zhang, M.Y.; Li, H.; Qu, Y.Q. Methylation and transcriptome analysis reveal lung adenocarcinoma-specific diagnostic biomarkers. J. Transl. Med. 2019, 17, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Dong, Q.; Li, F.; Xu, Y.; Hu, C.; Wang, J.; Shang, D.; Zheng, X.; Yang, H.; Zhang, C.; et al. Identifying subpathway signatures for individualized anticancer drug response by integrating multi-omics data. J. Transl. Med. 2019, 17, 255. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Song, C.; Ning, Z.; Ai, B.; Wang, Q.; Xu, Y.; Li, M.; Bai, X.; Zhao, J.; Liu, Y.; et al. ce-Subpathway: Identification of ceRNA-mediated subpathways via joint power of ceRNAs and pathway topologies. J. Cell. Mol. Med. 2019, 23, 967–984. [Google Scholar] [CrossRef]
- Sheng, Y.; Jiang, Y.; Yang, Y.; Li, X.; Qiu, J.; Wu, J.; Cheng, L.; Han, J. CNA2Subpathway: Identification of dysregulated subpathway driven by copy number alterations in cancer. Brief. Bioinform. 2021, 22, bbaa413. [Google Scholar] [CrossRef]
- Yang, E.V.; Sood, A.K.; Chen, M.; Li, Y.; Eubank, T.D.; Marsh, C.B.; Jewell, S.; Flavahan, N.A.; Morrison, C.; Yeh, P.-E.; et al. Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells. Cancer Res. 2006, 66, 10357–10364. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Han, X.; Kong, Q.; Cheng, L. psSubpathway: A software package for flexible identification of phenotype-specific subpathways in cancer progression. Bioinformatics 2020, 36, 2303–2305. [Google Scholar] [CrossRef]
- Han, J.; Liu, S.; Jiang, Y.; Xu, C.; Zheng, B.; Jiang, M.; Yang, H.; Su, F.; Li, C.; Zhang, Y. Inference of patient-specific subpathway activities reveals a functional signature associated with the prognosis of patients with breast cancer. J. Cell. Mol. Med. 2018, 22, 4304–4316. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, Y.; Ji, H.; Aihara, K.; Chen, L. Personalized characterization of diseases using sample-specific networks. Nucleic Acids Res. 2016, 44, e164. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Andorf, S.; Gomes, L.; Dunn, P.; Schaefer, H.; Pontius, J.; Berger, P.; Desborough, V.; Smith, T.; Campbell, J.; et al. ImmPort: Disseminating data to the public for the future of immunology. Immunol. Res. 2014, 58, 234–239. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Li, S.; Li, H.; Cao, Y.; Geng, H.; Ren, F.; Li, K.; Dai, C.; Li, N. Integrated bioinformatics analysis reveals CDK1 and PLK1 as potential therapeutic targets of lung adenocarcinoma. Medicine 2021, 100, e26474. [Google Scholar] [CrossRef]
- Li, S.; Jiang, Z.; Li, Y.; Xu, Y. Prognostic significance of minichromosome maintenance mRNA expression in human lung adenocarcinoma. Oncol. Rep. 2019, 42, 2279–2292. [Google Scholar] [CrossRef]
- Cheung, C.H.Y.; Hsu, C.L.; Chen, K.P.; Chong, S.T.; Wu, C.H.; Huang, H.C.; Juan, H.F. MCM2-regulated functional networks in lung cancer by multi-dimensional proteomic approach. Sci. Rep. 2017, 7, 13302. [Google Scholar] [CrossRef]
- Yi, J.; Wei, X.; Li, X.; Wan, L.; Dong, J.; Wang, R. A genome-wide comprehensive analysis of alterations in driver genes in non-small-cell lung cancer. Anti-Cancer Drugs 2018, 29, 10–18. [Google Scholar] [CrossRef]
- Kikuchi, J.; Kinoshita, I.; Shimizu, Y.; Kikuchi, E.; Takeda, K.; Aburatani, H.; Oizumi, S.; Konishi, J.; Kaga, K.; Matsuno, Y.; et al. Minichromosome maintenance (MCM) protein 4 as a marker for proliferation and its clinical and clinicopathological significance in non-small cell lung cancer. Lung Cancer 2011, 72, 229–237. [Google Scholar] [CrossRef]
- Tachibana, K.E.K.; Gonzalez, K.A.; Coleman, N. Cell-cycle-dependent regulation of DNA replication and its relevance to cancer pathology. J. Pathol. 2005, 205, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Vigouroux, C.; Casse, J.M.; Battaglia-Hsu, S.F.; Brochin, L.; Luc, A.; Paris, C.; Lacomme, S.; Gueant, J.L.; Vignaud, J.M.; Gauchotte, G. Methyl(R217) HuR and MCM6 are inversely correlated and are prognostic markers in non small cell lung carcinoma. Lung Cancer 2015, 89, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Shih, D.J.H.; Nayyar, N.; Bihun, I.; Dagogo-Jack, I.; Gill, C.M.; Aquilanti, E.; Bertalan, M.; Kaplan, A.; D’Andrea, M.R.; Chukwueke, U.; et al. Genomic characterization of human brain metastases identifies drivers of metastatic lung adenocarcinoma. Nat. Genet. 2020, 52, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Miyoshi, Y.; Takahata, C.; Irahara, N.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Down-regulation of LATS1 and LATS2 mRNA expression by promoter hypermethylation and its association with biologically aggressive phenotype in human breast cancers. Clin. Cancer Res. 2005, 11, 1380–1385. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, H. The Hippo signaling pathway components Lats and Yap pattern Tead4 activity to distinguish mouse trophectoderm from inner cell mass. Mech. Dev. 2009, 126, S22. [Google Scholar] [CrossRef]
- Zhang, J.; Smolen, G.A.; Haber, D.A. Negative regulation of YAP by LATS1 underscores evolutionary conservation of the Drosophila Hippo pathway. Cancer Res. 2008, 68, 2789–2794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.J.; Roumeliotis, T.I.; Chang, Y.H.; Chen, C.T.; Han, C.L.; Lin, M.H.; Chen, H.W.; Chang, G.C.; Chang, Y.L.; Wu, C.T.; et al. Proteogenomics of Non-smoking Lung Cancer in East Asia Delineates Molecular Signatures of Pathogenesis and Progression. Cell 2020, 182, 226–244. [Google Scholar] [CrossRef]
- Xu, J.Y.; Zhang, C.; Wang, X.; Zhai, L.; Ma, Y.; Mao, Y.; Qian, K.; Sun, C.; Liu, Z.; Jiang, S.; et al. Integrative Proteomic Characterization of Human Lung Adenocarcinoma. Cell 2020, 182, 245–261. [Google Scholar] [CrossRef]
- Lin, J.J.; Shaw, A.T. Resisting Resistance: Targeted Therapies in Lung Cancer. Trends Cancer 2016, 2, 350–364. [Google Scholar] [CrossRef] [Green Version]
- Peters, S.; Kerr, K.M.; Stahel, R. PD-1 blockade in advanced NSCLC: A focus on pembrolizumab. Cancer Treat. Rev. 2018, 62, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.; Liu, S.V.; Subramaniam, D.S.; Giaccone, G. Refining the treatment of NSCLC according to histological and molecular subtypes. Nat. Rev. Clin. Oncol. 2015, 12, 511–526. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Pian, C.; Xu, M.; Kong, L.; Li, Z.; Ji, J.; Zhang, L.; Chen, Y. Revealing Prognosis-Related Pathways at the Individual Level by a Comprehensive Analysis of Different Cancer Transcription Data. Genes 2020, 11, 1281. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enriched Pathways | The Number of Enriched Samples |
|---|---|
| Pyrimidine metabolism | 439 |
| Alcoholism | 394 |
| Cell cycle | 386 |
| Thyroid hormone signaling pathway | 386 |
| ECM–receptor interaction | 380 |
| Oxidative phosphorylation | 363 |
| Hippo signaling pathway | 360 |
| Inositol phosphate metabolism | 358 |
| Apoptosis | 356 |
| Phosphatidylinositol signaling system | 355 |
| Small cell lung cancer | 353 |
| Wnt signaling pathway | 344 |
| Pathways in cancer | 322 |
| Basal cell carcinoma | 309 |
| Lysine degradation | 301 |
| Focal adhesion | 300 |
| Axon guidance | 286 |
| PI3K-Akt signaling pathway | 284 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, J.; Li, Z.; Xu, M.; Ji, J.; Li, Y.; Zhang, L.; Chen, Y. Identification and Functional Analysis of Individual-Specific Subpathways in Lung Adenocarcinoma. Genes 2022, 13, 1122. https://doi.org/10.3390/genes13071122
Fang J, Li Z, Xu M, Ji J, Li Y, Zhang L, Chen Y. Identification and Functional Analysis of Individual-Specific Subpathways in Lung Adenocarcinoma. Genes. 2022; 13(7):1122. https://doi.org/10.3390/genes13071122
Chicago/Turabian StyleFang, Jingya, Zutan Li, Mingmin Xu, Jinwen Ji, Yanru Li, Liangyun Zhang, and Yuanyuan Chen. 2022. "Identification and Functional Analysis of Individual-Specific Subpathways in Lung Adenocarcinoma" Genes 13, no. 7: 1122. https://doi.org/10.3390/genes13071122
APA StyleFang, J., Li, Z., Xu, M., Ji, J., Li, Y., Zhang, L., & Chen, Y. (2022). Identification and Functional Analysis of Individual-Specific Subpathways in Lung Adenocarcinoma. Genes, 13(7), 1122. https://doi.org/10.3390/genes13071122