
Clinical and Mutation Spectrum of Autosomal Recessive Non-Syndromic Oculocutaneous Albinism (nsOCA) in Pakistan: A Review


Abstract
:1. Introduction
2. Literature Review
2.1. OCA Albinism
2.2. Melanin Biosynthesis Pathway and Its Link to OCA Types
2.3. Prevalence/Epidemiology
2.4. Phenotype Variations in OCA
3. Clinical Features of Different OCA Types
4. Molecular Classification of Genes with Non-Syndromic OCA
4.1. TYR (OCA1)
4.2. OCA2 (OCA2)
4.3. TYRP1 (OCA3)
4.4. SLC45A2 (OCA4)
4.5. OCA5
4.6. SLC24A5 (OCA6)
4.7. C10ORF11 (OCA7)
4.8. DCT Associated with OCA8 (New Type)
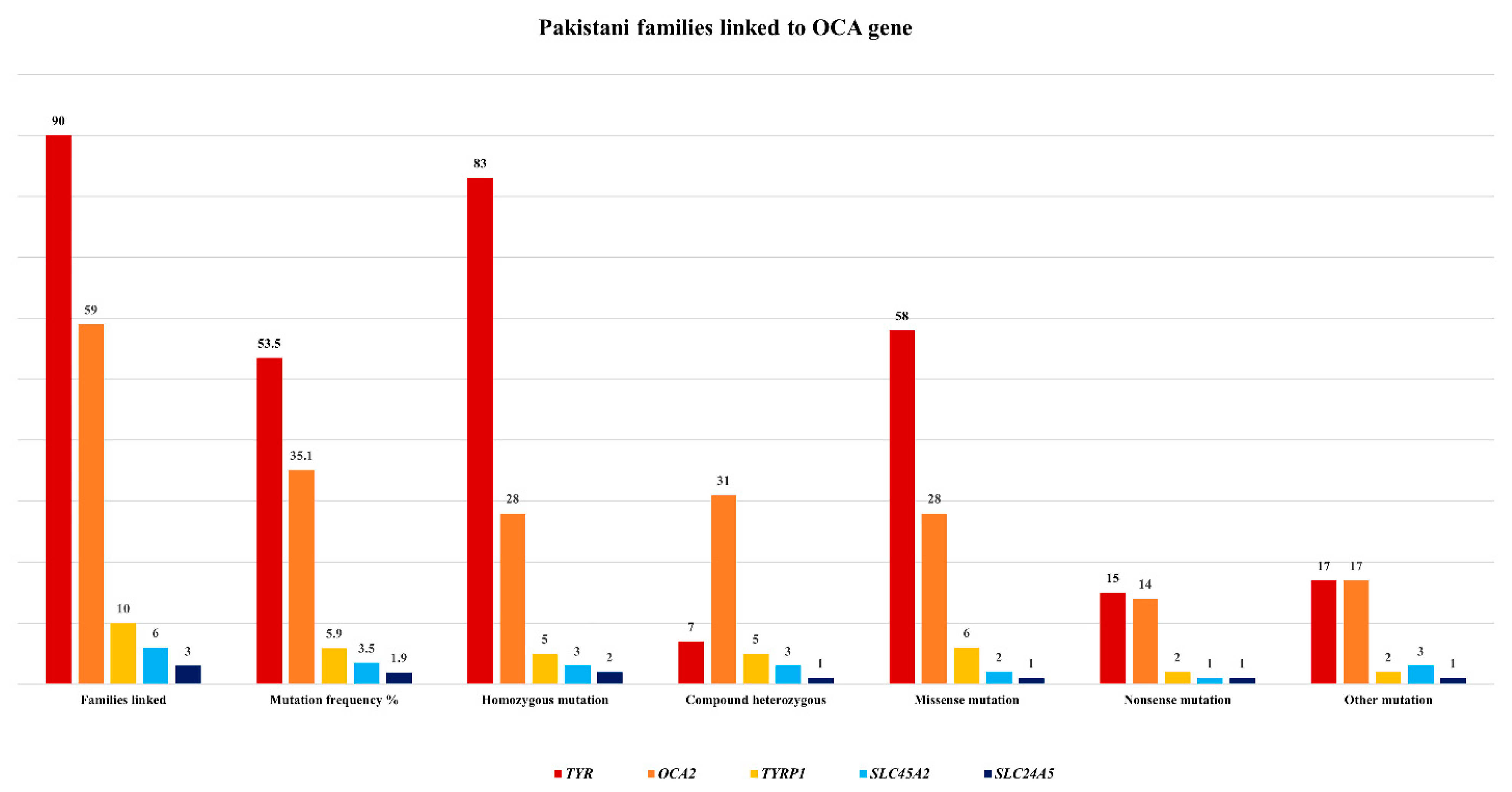
5. Mutation Spectrum of OCA Genes in the Pakistani Population

{kind=link}
{kind=link}
| Mutation | Language/Ethnic Group | Mutation Type and Status | No. Pakistani Families Linked to OCA | References |
|---|---|---|---|---|
| c.62 C>T; p.Pro21Leu | Punjabi | Missense/homozygous | 1 | [49] |
| c.103 T>C; p.Cys35Arg | Punjabi | Missense/homozygous | 2 | |
| c.132T>A; p.Ser44Arg | Kashmiri, Pakhtoon, Baluchi | Missense/homozygous | 3 | [9,81] |
| c.164G>C; p.Cys55Ser | Punjabi | Missense/homozygous | 1 | [64] |
| c.223G>T; p.Asp75Tyr | Punjabi | Missense/homozygous | 1 | |
| c.230G>A; p.Arg77Gln | Kashmiri | Missense/homozygous | 4 | [63] |
| c.248T G; p.Val83Gly | Pakhtoon | Missense/homozygous | 1 | [61] |
| c.240G>C; p.Trp80Cys | Pakhtoon | Missense/homozygous | 2 | [9] |
| c.272G>A; p.Cys91Tyr | Punjabi | Missense/homozygous | 1 | [65] |
| c.308G>A; p.Cys103Tyr | Punjabi | Missense/homozygous | 1 | |
| c.346C>T; p.Arg116 * | Saraiki, Punjabi | Nonsense/homozygous | 2 | [60,65] |
| c.575C>A; p.Ser192Tyr | Kashmiri | Missense/homozygous | 1 | |
| c.585G>A; p.Trp195 * | Punjabi | Nonsense/homozygous | 1 | [64] |
| c.593 T>C; p.Ile198Thr | Pakistani | Missense/homozygous | 1 | [62] |
| c.715C>T; p.Arg239Trp | Kashmiri | Missense/homozygous | 1 | [63] |
| c.832C>T; p.Arg278 * | Urdu speaking, Punjabi, Pakhtoon | Nonsense/homozygous | 21 | [9,21,42,49,63,64,65] |
| c.826T>C; p.Cys276Arg | Pakhtoon | Missense/homozygous | 1 | [42] |
| c.895C>T; p.Arg299Cys | Saraiki | Missense/homozygous | 1 | [61] |
| c.896A>G; p.Arg299His | Punjabi, Pakhtoon | Missense/homozygous | 3 | [56,64] |
| c.943–948delTCAGCT; p.315–316delSerAla | Punjabi | Deletion/heterozygous | 1 | [64] |
| c.982G>C; p.Glu328Gln | Pakistani origin (language/ethnicity not documented) | Missense/homozygous | 1 | [36] |
| c.1037 G>A; p.Gly346Glu | Punjabi | Missense/homozygous | 1 | [64] |
| c.1037G>T; p.Gly346Val | Punjabi | Missense/homozygous | 1 | |
| c.1037–7 T>A | Punjabi | Splicing error/heterozygous | 4 | |
| c.1037–18 T>G | Urdu speaking | Splicing error/heterozygous | 1 | |
| c.1147 G>A; p.Asp383Asn | Pakhtoon | Missense/homozygous | 1 | |
| c.115 del 340-341;frame shift p.R278X | Pakistani | Fame shift nonsense | 1 | [59] |
| c.1184 + 2 T>C | Punjabi | Splicing error/heterozygous | 1 | [64] |
| c.1204 C>T; p.Arg402 * | Punjabi | Nonsense/homozygous | 1 | |
| c.1217 C>T; p.Pro406Leu | Punjabi | Missense/homozygous | 1 | [64] |
| c.1231 T>C; p.Tyr411His | Punjabi | Missense/homozygous | 1 | |
| c.1255G>A; p.Gly419Arg | Punjabi, Pakhtoon, Sindhi, Kashmiri, Saraiki | Missense/homozygous | 23 | [9,21,49,60,61,63,64,65,81] |
| c.1424G>A; p.Trp475 * | Saraiki | Nonsense/homozygous | 1 | [61] |
| Exons 4–5 deletion | Punjabi | Deletion/heterozygous | 2 | [64] |
| Total families linked to TYR | Ninety | |||
| Mutation | Language/Ethnic Group | Mutation Type and Status | No. Pakistani Families Linked to OCA | References |
|---|---|---|---|---|
| c.827T>A; p.Val276Glu; c.877G>C; p.Glu293Gln | Kashmiri | Missense/compound heterozygous | 1 | [21] |
| TYR-c.832C>T; p.Arg278 *; OCA2-c.954G>A; p.Met318Ile | Sindhi | Missense/non-sense digenic/compound heterozygous | 1 | |
| TYR-c.649C>T; p.Arg217Trp; OCA2-c.1456G>T; p.Asp486Tyr | Sindhi | Missense digenic/compound heterozygous | 1 | |
| TYR-c.1255G>A; p.Gly419Arg; OCA2-c.954G>A; p.Met318Ile | Punjabi | Missense digenic/compound heterozygous | 1 | |
| c.2079 + 5G>T | Pakhtoon | Splice site/compound heterozygous | 1 | [40] |
| Ex19 del | Pakhtoon | Frame-shift/homozygous | 2 | [61] |
| c.1327G>A; p.Val443Ile; c.1762C>T; p.Arg588Trp | Punjabi | Missense/compound heterozygous | 1 | [9] |
| Exons 3–14 deletion | Punjabi | Frame-shift/homozygous | 1 | [64] |
| Exons 7–8 deletion | Punjabi | Frame-shift/homozygous | 2 | |
| c.1045–15 T>G | Punjabi, Saraiki, Sindhi | Splice site/compound heterozygous | 17 | [9,49,64] |
| c.1056 A>C | Sindhi | Missense/homozygous | 1 | [64] |
| c.1064 C>T | Sindhi | Missense/homozygous | 1 | |
| c.1075 G>C | Saraiki | Missense/homozygous | 1 | |
| c.1182 + 2 T>TT | Punjabi | Splice site/compound heterozygous | 1 | |
| c.1211 C>T | Punjabi | Missense/homozygous | 1 | |
| c.1322 A>G | Sindhi | Missense/homozygous | 1 | |
| c.1456 G>T | Punjabi, Saraiki, Sindhi | Missense/homozygous | 11 | [49,64] |
| c.1922 C>T | Punjabi | Missense/homozygous | 1 | [64] |
| c.1951 + 4 A>G | Urdu speaking | Splice site/compound heterozygous | 1 | |
| Exon 19 deletion | Punjabi | Deletion/compound heterozygous | 1 | |
| Exons 20–24 deletion | Pakhtoon, Punjabi | Deletion/compound heterozygous | 2 | |
| c.2020C>G; p.Leu674Val.; c.408_409delTT; p.Arg137Ilefs * 83 | Pakhtoon | Missense/frame shift/compound heterozygous | 1 | [9] |
| c.2228 C>T; p.Pro743Leu | Urdu speaking, Punjabi | Missense/homozygous | 2 | [49,64] |
| c.954 G>A; p.Met318Ile | Punjabi | Missense/homozygous | 1 | [49] |
| c.1580 T>G; p.Leu527Arg | Punjabi | Missense/homozygous | 1 | |
| c.2359 G>A; p.Ala787Thr | Punjabi | Missense/homozygous | 1 | |
| c.2360 C>A; p.Thr787Arg | Punjabi | Missense/homozygous | 1 | |
| c.2360 C>T; p.Thr787Ile | Punjabi | Missense/homozygous | 1 | |
| c.2458T>C; p.Ser820Pro | Punjabi | Missense/homozygous | 1 | [9] |
| Total families linked to OCA2 | Fifty nine | |||
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baulier, E.; Garcia Diaz, A.; Corneo, B.; Farber, D.B. Generation of a human Ocular Albinism type 1 iPSC line, SEIi001-A, with a mutation in GPR143. Stem. Cell. Res. 2018, 33, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Hamid, M.A.; Mehta, M.C.; Kuppermann, B.D. Multimodal imaging in a patient with Prader-Willi syndrome. Int. J. Retin. Vitr. 2018, 4, 45. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Oh, E.H.; Shin, J.H.; Kim, H.S.; Choi, S.Y.; Choi, K.D.; Lee, C.; Choi, J.H. Identification of a novel GPR143 mutation in X-linked ocular albinism with marked intrafamilial phenotypic variability. J. Genet. 2018, 97, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Garcia Galvão, L.E.; Tomaz, R.; de Sá Gonçalves, H. Daylight Photodynamic Therapy in the Treatment of Actinic Keratosis in Carriers of Oculocutaneous Albinism: Report of Three Cases. Actas Dermosifiliogr. 2019, 110, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Han, C.G.; O’Brien, K.J.; Coon, L.M.; Majerus, J.A.; Huryn, L.A.; Haroutunian, S.G.; Moka, N.; Introne, W.J.; Macnamara, E.; Gahl, W.A.; et al. Severe bleeding with subclinical oculocutaneous albinism in a patient with a novel HPS6 missense variant. Am. J. Med. Genet. A 2018, 176, 2819–2823. [Google Scholar] [CrossRef]
- Teramae, A.; Kobayashi, Y.; Kunimoto, H.; Nakajima, K.; Suzuki, T.; Tsuruta, D.; Fukai, K. The Molecular Basis of Chemical Chaperone Therapy for Oculocutaneous Albinism Type 1A. J. Invest. Derm. 2019, 139, 1143–1149. [Google Scholar] [CrossRef]
- Oetting, W.S.; Gardner, J.M.; Fryer, J.P.; Ching, A.; Durham-Pierre, D.; King, R.A.; Brilliant, M.H. Mutations of the human P gene associated with Type II oculocutaneous albinism (OCA2). Mutations in brief no. 205. Online. Hum. Mutat 1998, 12, 434. [Google Scholar] [CrossRef]
- Kubasch, A.S.; Meurer, M. Okulokutaner und okulärer Albinismus [Oculocutaneous and ocular albinism]. Hautarzt 2017, 68, 867–875. [Google Scholar] [CrossRef]
- Arshad, M.W.; Harlalka, G.V.; Lin, S.; D’Atri, I.; Mehmood, S.; Shakil, M.; Hassan, M.J.; Chioza, B.A.; Self, J.E.; Ennis, S.; et al. Mutations in TYR and OCA2 associated with oculocutaneous albinism in Pakistani families. Meta Gene 2018, 17, 48–55. [Google Scholar] [CrossRef]
- Dooleyl, C.M.; Schwarz, H.; Mueller, K.P.; Mongera, A.; Konantz, M.; Neuhauss, S.C.; Nüsslein-Volhard, C.; Geisler, R. Slc45a2 and V-ATPase are regulators of melanosomal pH homeostasis in zebrafish, providing a mechanism for human pigment evolution and disease. Pigment. Cell Melanoma Res. 2013, 26, 205–217. [Google Scholar] [CrossRef]
- Cordero, R.J.; Casadevall, A. Functions of fungal melanin beyond virulence. Fungal. Biol. Rev. 2017, 31, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Videira, I.F.; Moura, D.F.; Magina, S. Mechanisms regulating melanogenesis. An. Bras. Derm. 2013, 88, 76–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Mello, S.A.; Finlay, G.J.; Baguley, B.C.; Askarian-Amiri, M.E. Signaling Pathways in Melanogenesis. Int. J. Mol. Sci. 2016, 17, 1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costin, G.E.; Hearing, V.J. Human skin pigmentation: Melanocytes modulate skin color in response to stress. FASEB J. 2007, 21, 976–994. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Nicholls, R.D.; Schnur, R.E.; Guida, L.C.; Lu-Kuo, J.; Spinner, N.B.; Zackai, E.H.; Spritz, R.A. Diverse mutations of the P gene among African-Americans with type II (tyrosinase-positive) oculocutaneous albinism (OCA2). Hum. Mol. Genet. 1994, 3, 2047–2051. [Google Scholar]
- Manga, P.; Kromberg, J.; Turner, A.; Jenkins, T.; Ramsay, M. In Southern Africa, brown oculocutaneous albinism (BOCA) maps to the OCA2 locus on chromosome 15q: P-gene mutations identified. Am. J. Hum. Genet 2001, 68, 782–787. [Google Scholar] [CrossRef] [Green Version]
- Chaki, M.; Sengupta, M.; Mukhopadhyay, A.; Subba Rao, I.; Majumder, P.P.; Das, M.; Samanta, S.; Ray, K. OCA1 in different ethnic groups of india is primarily due to founder mutations in the tyrosinase gene. Ann. Hum. Genet. 2006, 70, 623–630. [Google Scholar] [CrossRef]
- Ghodsinejad Kalahroudi, V.; Kamalidehghan, B.; Arasteh Kani, A.; Aryani, O.; Tondar, M.; Ahmadipour, F.; Chung, L.Y.; Houshmand, M. Two novel tyrosinase (TYR) gene mutations with pathogenic impact on oculocutaneous albinism type 1 (OCA1). PLoS ONE 2014, 9, e106656. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Kong, X.D.; Shi, H.R.; Wu, Q.H.; Jiang, M. Tyrosinase gene mutations in the Chinese Han population with OCA1. Genet. Res. 2014, 96, e14. [Google Scholar] [CrossRef]
- Norman, C.S.; O’Gorman, L.; Gibson, J.; Pengelly, R.J.; Baralle, D.; Ratnayaka, J.A.; Griffiths, H.; Rose-Zerilli, M.; Ranger, M.; Bunyan, D.; et al. Identification of a functionally significant tri-allelic genotype in the Tyrosinase gene (TYR) causing hypomorphic oculocutaneous albinism (OCA1B). Sci. Rep. 2017, 7, 4415. [Google Scholar] [CrossRef]
- Sajid, Z.; Yousaf, S.; Waryah, Y.M.; Mughal, T.A.; Kausar, T.; Shahzad, M.; Rao, A.R.; Abbasi, A.A.; Shaikh, R.S.; Waryah, A.M.; et al. Genetic Causes of Oculocutaneous Albinism in Pakistani Population. Genes 2021, 1, 492. [Google Scholar] [CrossRef] [PubMed]
- Kausar, T.; Bhatti, M.A.; Ali, M.; Shaikh, R.S.; Ahmed, Z.M. OCA5, a novel locus for non-syndromic oculocutaneous albinism, maps to chromosome 4q24. Clin. Genet. 2013, 84, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Lasseaux, E.; Plaisant, C.; Trimouille, A.; Morice-Picard, F.; Rooryck, C.; Lacombe, D.; Couppie, P.; Arveiler, B. Identification of a homozygous mutation of SLC24A5 (OCA6) in two patients with oculocutaneous albinism from French Guiana. Pigment. Cell Melanoma Res. 2016, 29, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Veniani, E.; Mauri, L.; Manfredini, E.; Gesu, G.P.; Patrosso, M.C.; Zelante, L.; D’Agruma, L.; Del Longo, A.; Mazza, M.; Piozzi, E.; et al. Detection of the first OCA6 Italian patient in a large cohort of albino subjects. J. Derm. Sci. 2016, 81, 208–209. [Google Scholar] [CrossRef]
- Yousaf, S.; Sethna, S.; Chaudhary, M.A.; Shaikh, R.S.; Riazuddin, S.; Ahmed, Z.M. Molecular characterization of SLC24A5 variants and evaluation of Nitisinone treatment efficacy in a zebrafish model of OCA6. Pigment. Cell Melanoma Res. 2020, 33, 556–565. [Google Scholar] [CrossRef]
- Federico, J.R.; Krishnamurthy, K. Albinism; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Camand, O.; Marchant, D.; Boutboul, S.; Péquignot, M.; Odent, S.; Dollfus, H.; Sutherland, J.; Levin, A.; Menasche, M.; Marsac, C.; et al. Mutation analysis of the tyrosinase gene in oculocutaneous albinism. Hum. Mutat 2001, 17, 352. [Google Scholar] [CrossRef]
- Ganguly, K.; Dutta, T.; Saha, A.; Sarkar, D.; Sil, A.; Ray, K.; Sengupta, M. Mapping the TYR gene reveals novel and previously reported variants in Eastern Indian patients highlighting preponderance of the same changes in multiple unrelated ethnicities. Ann. Hum. Gene. 2020, 84, 303–312. [Google Scholar] [CrossRef]
- Giebel, L.B.; Tripathi, R.K.; Strunk, K.M.; Hanifin, J.M.; Jackson, C.E.; King, R.A.; Spritz, R.A. Tyrosinase gene mutations associated with type IB (yellow) oculocutaneous albinism. Am. J. Hum. Genet. 1991, 48, 1159–1167, Erratum in Am. J. Hum. Genet. 1991, 49, 696. [Google Scholar]
- Manga, P.; Kromberg, J.G.; Box, N.F.; Sturm, R.A.; Jenkins, T.; Ramsay, M. Rufous oculocutaneous albinism in southern African Blacks is caused by mutations in the TYRP1 gene. Am. J. Hum. Genet. 1997, 61, 1095–1101. [Google Scholar] [CrossRef] [Green Version]
- Rooryck, C.; Morice-Picard, F.; Elçioglu, N.H.; Lacombe, D.; Taieb, A.; Arveiler, B. Molecular diagnosis of oculocutaneous albinism: New mutations in the OCA1-4 genes and practical aspects. Pigment. Cell Melanoma Res. 2008, 21, 583–587. [Google Scholar] [CrossRef]
- Witkop, C.J.; Rao, G.; Gaudier, F.; Summers, C.; Shanahan, F.; Harmon, K.; Townsend, D.; Sedano, H.O.; King, P.A. Albinism and Hermansky-Pudlak syndrome in Puerto Rico. Bol. Asoc. Med. Puerto Rico 1990, 82, 333–339. [Google Scholar]
- Summers, C.G.; Knobloch, W.H.; Witkop, C.J.; King, R.A. Hermansky-Pudlak syndrome: Ophthalmic findings. Ophthalmology 1988, 95, 545–554. [Google Scholar] [CrossRef]
- Dorey, S.E.; Neveu, M.M.; Burton, L.C.; Sloper, J.J.; Holder, G.E. The clinical features of albinism and their correlation with visual evoked potentials. Br. J. Ophthalmol. 2003, 87, 767–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, R.K.; Bundey, S.; Musarella, M.A.; Droetto, S.; Strunk, K.M.; Holmes, S.A.; Spritz, R.A. Mutations of the tyrosinase gene in Indo-Pakistani patients with type I (tyrosinase-deficient) oculocutaneous albinism (OCA). Am. J. Hum. Genet. 1993, 53, 1173–1179. [Google Scholar] [PubMed]
- Lee, S.Y.; Lee, E.J.; Byun, J.C.; Jang, K.M.; Kim, S.Y.; Hwang, S.K. A new type of oculocutaneous albinism with a novel OCA2 mutation. Yeungnam Univ. J. Med. 2021, 38, 160–164. [Google Scholar] [CrossRef]
- Oetting, W.S.; Garrett, S.S.; Brott, M.; King, R.A. P gene mutations associated with oculocutaneous albinism type II (OCA2). Hum. Mutat 2005, 25, 323. [Google Scholar] [CrossRef]
- Okamura, K.; Hayashi, M.; Abe, Y.; Kono, M.; Nakajima, K.; Aoyama, Y.; Nishigori, C.; Ishimoto, H.; Ishimatsu, Y.; Nakajima, M.; et al. NGS-based targeted resequencing identified rare subtypes of albinism: Providing accurate molecular diagnosis for Japanese patients with albinism. Pigment. Cell Melanoma Res. 2019, 32, 848–853. [Google Scholar] [CrossRef]
- Okamura, K.; Suzuki, T. Current landscape of Oculocutaneous Albinism in Japan. Pigment. Cell Melanoma Res. 2021, 34, 190–203. [Google Scholar] [CrossRef]
- Zhang, K.H.; Li, Z.; Lei, J.; Pang, T.; Xu, B.; Jiang, W.Y.; Li, H.Y. Oculocutaneous albinism type 3 (OCA3): Analysis of two novel mutations in TYRP1 gene in two Chinese patients. Cell Biochem. Biophys. 2011, 61, 523–529. [Google Scholar] [CrossRef]
- Bibi, N.; Ullah, A.; Darwesh, L.; Khan, W.; Khan, T.; Ullah, K.; Khan, B.; Ahmad, W.; Umm-e-Kalsoom. Identification and Computational Analysis of Novel TYR and SLC45A2 Gene Mutations in Pakistani Families With Identical Non-syndromic Oculocutaneous Albinism. Front. Genet. 2020, 11, 749. [Google Scholar] [CrossRef]
- Konno, T.; Abe, Y.; Kawaguchi, M.; Storm, K.; Biervliet, M.; Courtens, W.; Kono, M.; Tomita, Y.; Suzuki, T. Oculocutaneous albinism type IV: A boy of Moroccan descent with a novel mutation in SLC45A2. Am. J. Med. Genet. A 2009, 149, 1773–1776. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, M.; Chaki, M.; Arti, N.; Ray, K. SLC45A2 variations in Indian oculocutaneous albinism patients. Mol. Vis. 2007, 13, 1406–1411. [Google Scholar] [PubMed]
- Wei, A.H.; Zang, D.J.; Zhang, Z.; Liu, X.Z.; He, X.; Yang, L.; Wang, Y.; Zhou, Z.Y.; Zhang, M.R.; Dai, L.L.; et al. Exome sequencing identifies SLC24A5 as a candidate gene for nonsyndromic oculocutaneous albinism. J. Investig. Derm. 2013, 133, 1834–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grønskov, K.; Dooley, C.M.; Østergaard, E.; Kelsh, R.N.; Hansen, L.; Levesque, M.P.; Vilhelmsen, K.; Møllgård, K.; Stemple, D.L.; Rosenberg, T. Mutations in c10orf11, a melanocyte-differentiation gene, cause autosomal-recessive albinism. Am. J. Hum. Genet. 2013, 92, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Pennamen, P.; Tingaud-Sequeira, A.; Gazova, I.; Keighren, M.; McKie, L.; Marlin, S.; Gherbi Halem, S.; Kaplan, J.; Delevoye, C.; Lacombe, D.; et al. Dopachrome tautomerase variants in patients with oculocutaneous albinism. Genet. Med. 2021, 23, 479–487. [Google Scholar] [CrossRef]
- Volk, A.E.; Hedergott, A.; Preising, M.; Rading, S.; Fricke, J.; Herkenrath, P.; Nürnberg, P.; Altmüller, J.; von Ameln, S.; Lorenz, B.; et al. Biallelic mutations in L-dopachrome tautomerase (DCT) cause infantile nystagmus and oculocutaneous albinism. Hum. Genet. 2022, 140, 1157–1168. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, S.M.; Im, K.R.; Yoon, K.S. Effect of hydroxycinnamic acid derivatives from corn bran on melanogenic protein expression. J. Korean Soc. Appl. Biol. Chem. 2010, 53, 422–426. [Google Scholar] [CrossRef]
- Shah, S.A.; Din, S.U.; Raheem, N.; Daud, S.; Mubeen, J.; Nadeem, A.; Tayyab, M.; Baloch, D.M.; Babar, M.E.; Ahmad, J. Identification of a novel mutation (p.Ile198Thr) in gene TYR in a Pakistani family with nonsyndromic oculocutaneous albinism. Clin. Exp. Derm. 2014, 39, 646–648. [Google Scholar] [CrossRef]
- Kromberg, J.G.; Bothwell, J.; Kidson, S.H.; Manga, P.; Kerr, R.; Jenkins, T. Types of Albinism in the Black Southern Africa Population. East. Afr. Med. J. 2012, 89, 20–27. [Google Scholar]
- Rooryck, C.; Roudaut, C.; Robine, E.; Müsebeck, J.; Arveiler, B. Oculocutaneous albinism with TYRP1 gene mutations in a Caucasian patient. Pigment. Cell Res. 2006, 19, 239–242. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Liu, T.; Bai, D.; Yang, X.; Li, W.; Wei, A. Identification of two Chinese oculocutaneous albinism type 6 patients and mutation updates of the SLC24A5 gene. J. Derm. 2019, 46, 1027–1030. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Sakai, K.; Hayashi, M.; Hozumi, Y.; Abe, Y.; Kawaguchi, M.; Ihn, H.; Suzuki, T. Oculocutaneous albinism type 3: A Japanese girl with novel mutations in TYRP1 gene. J. Derm. Sci. 2011, 64, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Ullah, F.; Khan, I.; Shah, S.A.; Azam, S.; Faisal, S.; Muzamil, H. Mutational Analysis of TYR Gene Causing Oculocutaneous Albinism in Families from District Peshawar. Int, J. Hum. Genet. 2019, 19, 170–178. [Google Scholar] [CrossRef]
- Morice-Picard, F.; Lasseaux, E.; François, S.; Simon, D.; Rooryck, C.; Bieth, E.; Colin, E.; Bonneau, D.; Journel, H.; Walraedt, S.; et al. SLC24A5 mutations are associated with non-syndromic oculocutaneous albinism. J. Investig. Derm. 2014, 134, 568–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simeonov, D.R.; Wang, X.; Wang, C.; Sergeev, Y.; Dolinska, M.; Bower, M.; Fischer, R.; Winer, D.; Dubrovsky, G.; Balog, J.Z.; et al. DNA variations in oculocutaneous albinism: An updated mutation list and current outstanding issues in molecular diagnostics. Hum. Mutat. 2013, 34, 827–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, R.A.; Pietsch, J.; Fryer, J.P.; Savage, S.; Brott, M.J.; Russell-Eggitt, I.; Summers, C.G.; Oetting, W.S. Tyrosinase gene mutations in oculocutaneous albinism 1 (OCA1): Definition of the phenotype. Hum. Genet. 2003, 113, 502–513. [Google Scholar] [CrossRef]
- Oetting, W.S. The tyrosinase gene and oculocutaneous albinism type 1 (OCA1): A model for understanding the molecular biology of melanin formation. Pigment. Cell Res. 2000, 13, 320–325. [Google Scholar] [CrossRef]
- Gul, H.; Ali, M.Z.; Khan, E.; Zubair, M.; Badar, M.; Khan, S.; Shah, A.H.; Khan, M.A. Ophthalmo-genetic analysis of Pakistani patients with nonsyndromic oculocutaneous albinism through whole exome sequencing. J. Pak. Med. Assoc. 2017, 67, 790–792. [Google Scholar]
- Gul, H.; Shah, A.H.; Harripaul, R.; Mikhailov, A.; Prajapati, K.; Khan, E.; Ullah, F.; Zubair, M.; Ali, M.Z.; Shah, A.H.; et al. Genetic studies of multiple consanguineous Pakistani families segregating oculocutaneous albinism identified novel and reported mutations. Ann. Hum. Genet. 2019, 83, 278–284. [Google Scholar] [CrossRef]
- Jaworek, T.J.; Kausar, T.; Bell, S.M.; Tariq, N.; Maqsood, M.I.; Sohail, A.; Ali, M.; Iqbal, F.; Rasool, S.; Riazuddin, S.; et al. Molecular genetic studies and delineation of the oculocutaneous albinism phenotype in the Pakistani population. Orphanet. J. Rare Dis. 2012, 7, 44. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.A.; Raheem, N.; Daud, S.; Mubeen, J.; Shaikh, A.A.; Baloch, A.H.; Nadeem, A.; Tayyab, M.; Babar, M.E.; Ahmad, J. Mutational spectrum of the TYR and SLC45A2 genes in Pakistani families with oculocutaneous albinism, and potential founder effect of missense substitution (p.Arg77Gln) of tyrosinase. Clin. Exp. Derm. 2015, 40, 774–780. [Google Scholar] [CrossRef]
- Shahzad, M.; Yousaf, S.; Waryah, Y.M.; Gul, H.; Kausar, T.; Tariq, N.; Mahmood, U.; Ali, M.; Khan, M.A.; Waryah, A.M.; et al. University of Washington Center for Mendelian Genomics (UW CMG) Consortium. Molecular outcomes, clinical consequences, and genetic diagnosis of Oculocutaneous Albinism in Pakistani population. Sci. Rep. 2017, 7, 44185. [Google Scholar] [CrossRef] [Green Version]
- Shakil, M.; Harlalka, G.V.; Ali, S.; Lin, S.; D’Atri, I.; Hussain, S.; Nasir, A.; Shahzad, M.A.; Ullah, M.I.; Self, J.E.; et al. Tyrosinase (TYR) gene sequencing and literature review reveals recurrent mutations and multiple population founder gene mutations as causative of oculocutaneous albinism (OCA) in Pakistani families. Eye 2019, 33, 1339–1346. [Google Scholar] [CrossRef]
- Spritz, R.A.; Fukai, K.; Holmes, S.A.; Luande, J. Frequent intragenic deletion of the P gene in Tanzanian patients with type II oculocutaneous albinism (OCA2). Am. J. Hum. Genet. 1995, 56, 1320–1323. [Google Scholar]
- Preising, M.N.; Forster, H.; Tan, H.; Lorenz, B.; de Jong, P.T.; Plomp, A.S. Mutation analysis in a family with oculocutaneous albinism manifesting in the same generation of three branches. Mol. Vis. 2007, 13, 1851–1855. [Google Scholar]
- Chen, Y.; Samaraweera, P.; Sun, T.T.; Kreibich, G.; Orlow, S.J. Rab27b association with melanosomes: Dominant negative mutants disrupt melanosomal movement. J. Investig. Derm. 2002, 118, 933–940. [Google Scholar] [CrossRef] [Green Version]
- Kamaraj, B.; Purohit, R. Mutational analysis of oculocutaneous albinism: A compact review. Biomed. Res. Int. 2014, 2014, 905472. [Google Scholar] [CrossRef] [Green Version]
- Khordadpoor-Deilamani, F.; Akbari, M.T.; Karimipoor, M.; Javadi, G.R. Homozygosity mapping in albinism patients using a novel panel of 13 STR markers inside the nonsyndromic OCA genes: Introducing 5 novel mutations. J. Hum. Genet. 2016, 61, 373–379. [Google Scholar] [CrossRef]
- Gul, H.; Shah, A.H.; Harripaul, R.; Mikhailov, A.; Khan, E.U.; Shah, W.; Ahmad, N.; Vincent, J.B.; Khan, M.A. Mutation Analysis of a Pakistani Oculocutaneous Albinism Family Identifies a Novel Splice Site Defect in OCA2 Gene. Pak. J. Zool. 2022, 54, 1215–1221. [Google Scholar] [CrossRef]
- Forshew, T.; Khaliq, S.; Tee, L.; Smith, U.; Johnson, C.A.; Mehdi, S.Q.; Maher, E.R. Identification of novel TYR and TYRP1 mutations in oculocutaneous albinism. Clin. Genet. 2005, 68, 182–184. [Google Scholar] [CrossRef]
- Inagaki, K.; Suzuki, T.; Ito, S.; Suzuki, N.; Adachi, K.; Okuyama, T.; Nakata, Y.; Shimizu, H.; Matsuura, H.; Oono, T.; et al. Oculocutaneous albinism type 4: Six novel mutations in the membrane-associated transporter protein gene and their phenotypes. Pigment. Cell Res. 2006, 19, 451–453. [Google Scholar] [CrossRef]
- Zhong, Z.; Gu, L.; Zheng, X.; Ma, N.; Wu, Z.; Duan, J.; Zhang, J.; Chen, J. Comprehensive analysis of spectral distribution of a large cohort of Chinese patients with non-syndromic oculocutaneous albinism facilitates genetic diagnosis. Pigment. Cell Melanoma Res. 2019, 32, 672–686. [Google Scholar] [CrossRef]
- Tzschach, A.; Bisgaard, A.M.; Kirchhoff, M.; Graul-Neumann, L.M.; Neitzel, H.; Page, S.; Ahmed, A.; Müller, I.; Erdogan, F.; Ropers, H.H.; et al. Chromosome aberrations involving 10q22: Report of three overlapping interstitial deletions and a balanced translocation disrupting C10orf11. Eur. J. Hum. Genet. 2010, 18, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, K.; Dutta, T.; Samanta, S.; Sil, A.; Ray, K. C10ORF11 is unlikely to have a major contribution towards OCA pathogenesis in Southern and Eastern India. J. Hum. Biol. Health Edu. 2017, 1, 1. [Google Scholar]
- Garrido, G.; Fernández, A.; Montoliu, L. HPS11 and OCA8: Two new types of albinism associated with mutations in BLOC1S5 and DCT genes. Pigment. Cell Melanoma Res. 2021, 34, 10–12. [Google Scholar] [CrossRef]
- Sendoel, A.; Kohler, I.; Fellmann, C.; Lowe, S.W.; Hengartner, M.O. HIF-1 antagonizes p53-mediated apoptosis through a secreted neuronal tyrosinase. Nature 2010, 465, 577–583. [Google Scholar] [CrossRef] [Green Version]
- Anwar, I.; Hussain, S.; Rehman, A.U.; Hussain, M. Genetic variation among the major Pakistani populations based on 15 autosomal STR markers. Int. J. Leg. Med. 2019, 133, 1037–1038. [Google Scholar] [CrossRef]
- King, R.A.; Willaert, R.K.; Schmidt, R.M.; Pietsch, J.; Savage, S.; Brott, M.J.; Fryer, J.P.; Summers, C.G.; Oetting, W.S. MC1R mutations modify the classic phenotype of oculocutaneous albinism type 2 (OCA2). Am. J. Hum. Genet. 2003, 73, 638–645. [Google Scholar] [CrossRef] [Green Version]
- Hutton, S.M.; Spritz, R.A. Comprehensive analysis of oculocutaneous albinism among non-Hispanic caucasians shows that OCA1 is the most prevalent OCA type. J. Investig. Derm. 2008, 128, 2442–24450. [Google Scholar] [CrossRef] [Green Version]
- Shakil, M.; Akbar, A.; Aisha, N.M.; Hussain, I.; Ullah, M.I.; Atif, M.; Kaul, H.; Amar, A.; Latif, M.Z.; Qureshi, M.A.; et al. Delineating Novel and Known Pathogenic Variants in TYR, OCA2 and HPS-1 Genes in Eight Oculocutaneous Albinism (OCA) Pakistani Families. Genes 2022, 13, 503. [Google Scholar] [CrossRef]
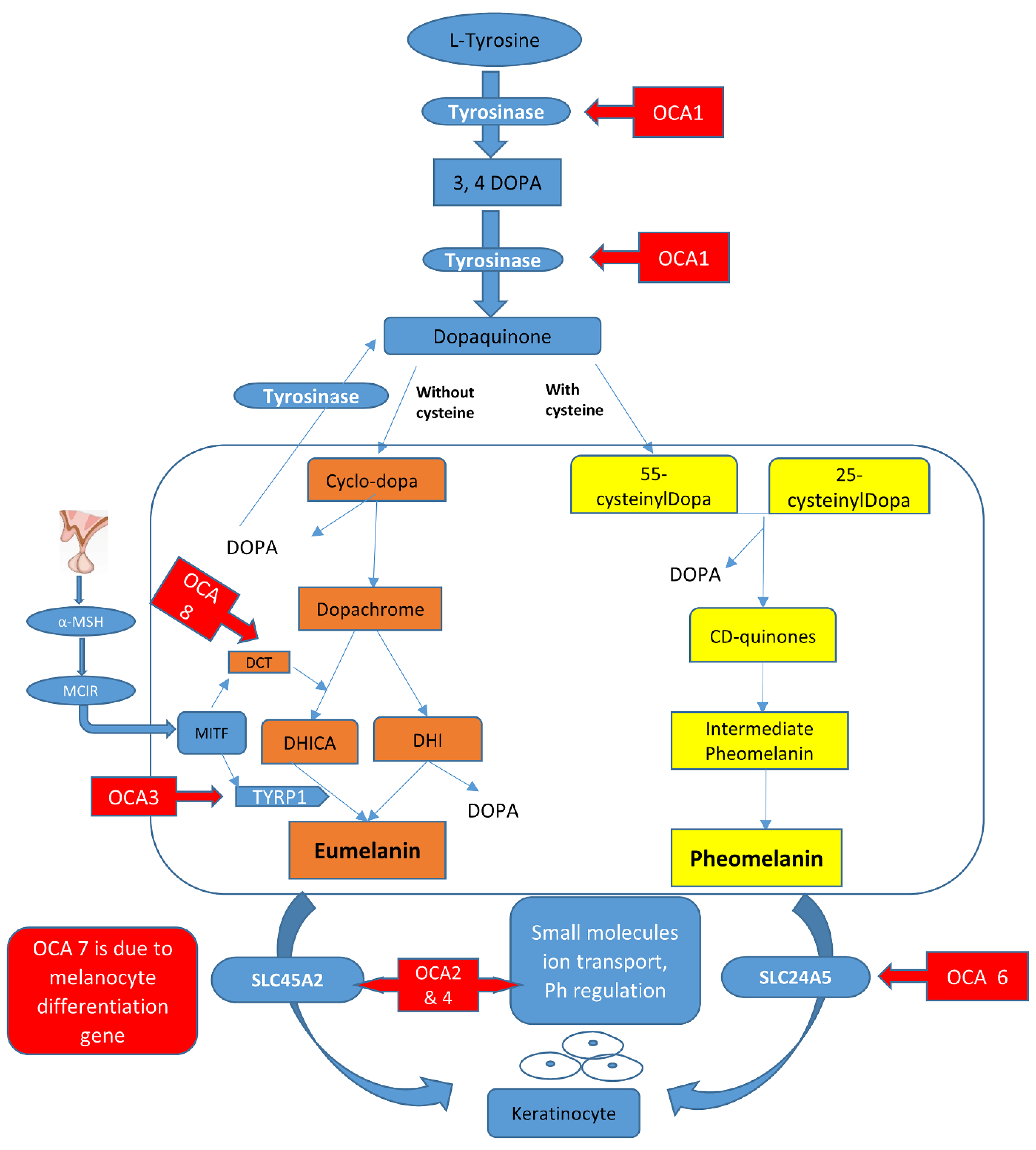
| Locus Name | Physical Location | Causative Gene | Cellular Location | Proposed Function | Reported Mutations | References |
|---|---|---|---|---|---|---|
| OCA1 | 11q14 | TYR | Melanosome | Malanogenesis | 487 | [42,49,50] |
| OCA2 | 15q11 | OCA2 | Transmembrane of melanosome | Regulate and transport tyrosinase | 364 | [51] |
| OCA3 | 9p23 | TYRP1 | Melanosome | Stabilize tyrosinase | 66 | [52,53,54] |
| OCA4 | 5p13 | SLC45A2 | Transmembrane of melanosome | Maintain melanosomal pH and help in binding of copper to tyrosinase | 86 | [40,55] |
| OCA5 | 4q24 | - | - | - | - | [22] |
| OCA6 | 15q21 | SLC24A5 | Melanosome | Ion exchange in melanocytes | 37 | [22,45] |
| OCA7 | 10q22 | C10orf11 | Melanocyte | Melanocyte Differentiation | 6 | [46] |
| OCA8 | 13q31 | DCT | Melanocyte | Melanosome apoptosis | 4 | [47,48] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, M.I. Clinical and Mutation Spectrum of Autosomal Recessive Non-Syndromic Oculocutaneous Albinism (nsOCA) in Pakistan: A Review. Genes 2022, 13, 1072. https://doi.org/10.3390/genes13061072
Ullah MI. Clinical and Mutation Spectrum of Autosomal Recessive Non-Syndromic Oculocutaneous Albinism (nsOCA) in Pakistan: A Review. Genes. 2022; 13(6):1072. https://doi.org/10.3390/genes13061072
Chicago/Turabian StyleUllah, Muhammad Ikram. 2022. "Clinical and Mutation Spectrum of Autosomal Recessive Non-Syndromic Oculocutaneous Albinism (nsOCA) in Pakistan: A Review" Genes 13, no. 6: 1072. https://doi.org/10.3390/genes13061072
APA StyleUllah, M. I. (2022). Clinical and Mutation Spectrum of Autosomal Recessive Non-Syndromic Oculocutaneous Albinism (nsOCA) in Pakistan: A Review. Genes, 13(6), 1072. https://doi.org/10.3390/genes13061072