
Assembly of a Large Collection of Maxicircle Sequences and Their Usefulness for Leishmania Taxonomy and Strain Typing

 , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Genomic Sequences and Data Availability
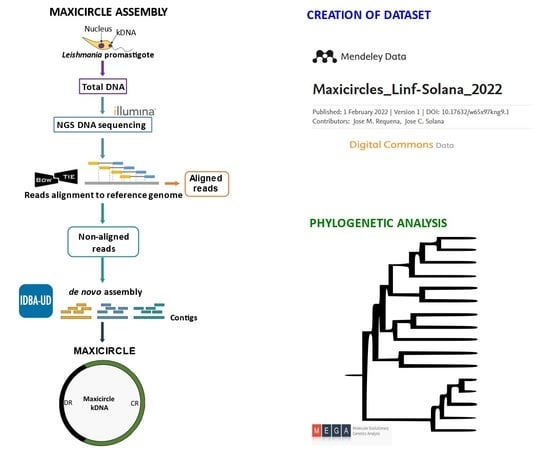
2.2. Assembly of Maxicircle Sequences
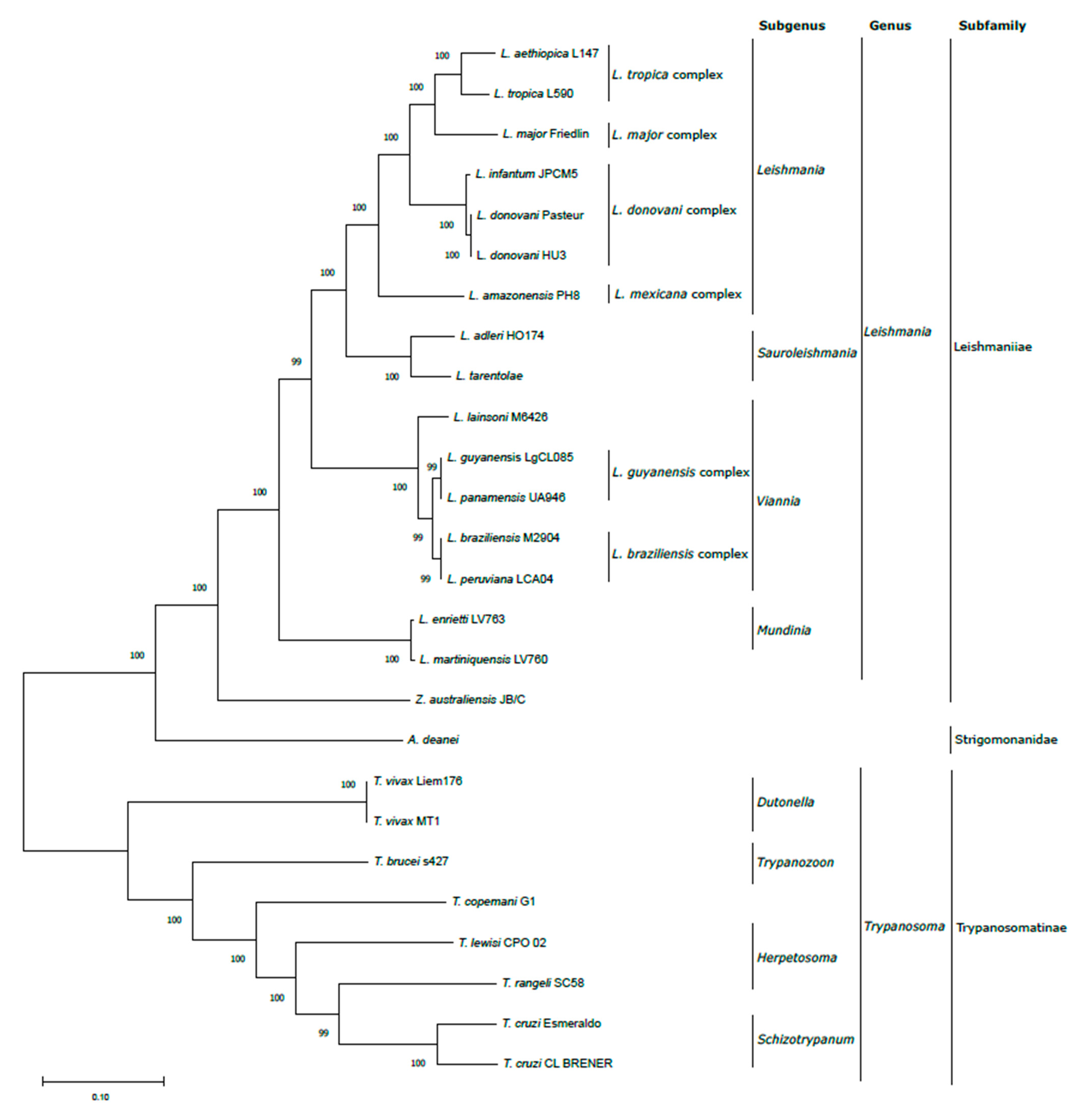
2.3. Phylogenetic Analysis
3. Results and Discussion
3.1. Creation of a Maxicircle Dataset for Trypanosomatids
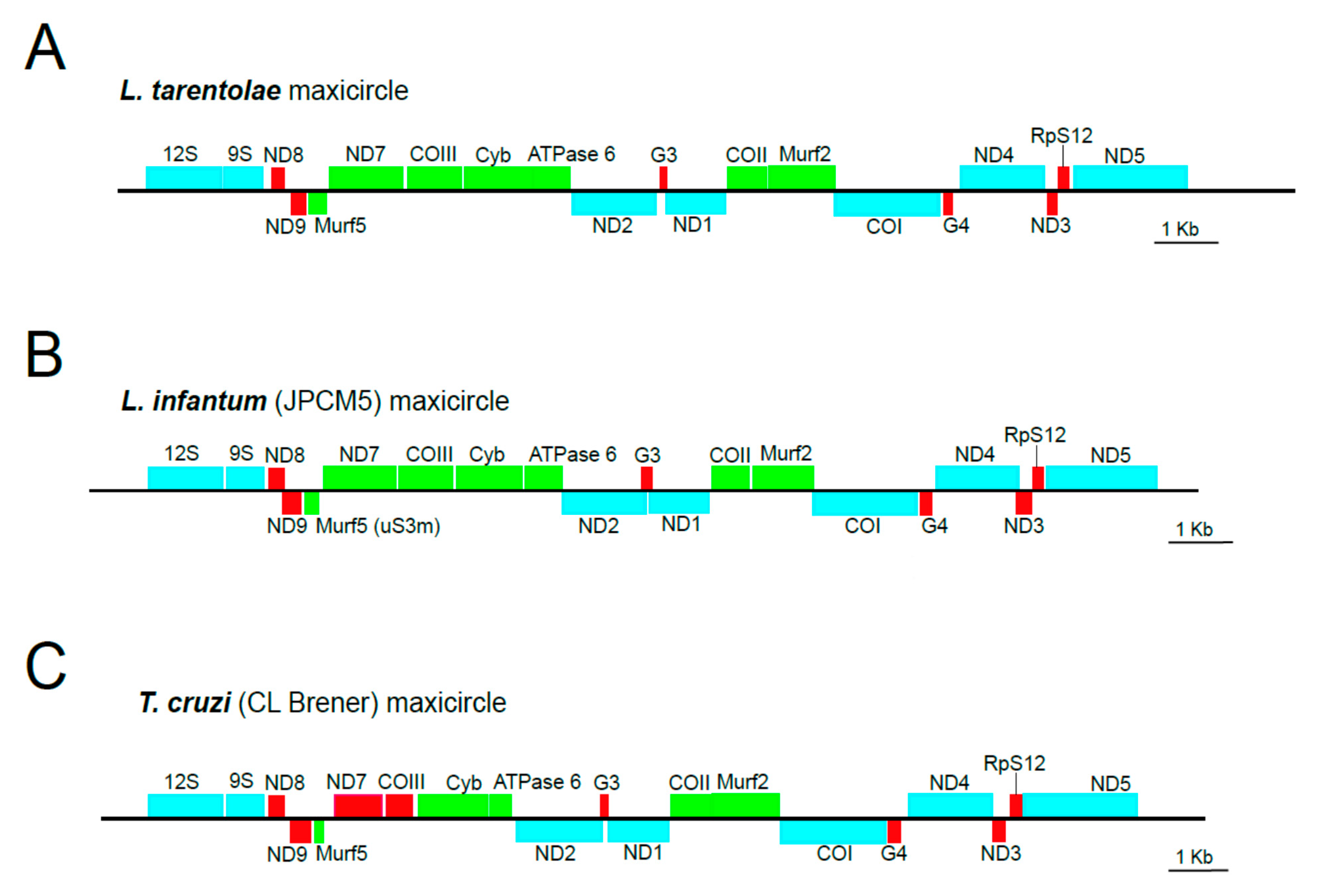
3.2. Validation of In Silico Assembled Maxicircle CR for Species Identification and Phylogenetic Analyses
3.3. Validation of In Silico Assembled Maxicircle CR for Species Identification and Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Volpedo, G.; Huston, R.H.; Holcomb, E.A.; Pacheco-Fernandez, T.; Gannavaram, S.; Bhattacharya, P.; Nakhasi, H.L.; Satoskar, A.R. From infection to vaccination: Reviewing the global burden, history of vaccine development, and recurring challenges in global leishmaniasis protection. Expert Rev. Vaccines 2021, 20, 1431–1446. [Google Scholar] [CrossRef] [PubMed]
- Moretti, N.S.; Mortara, R.A.; Schenkman, S. Trypanosoma cruzi. Trends Parasitol. 2020, 36, 404–405. [Google Scholar] [CrossRef] [PubMed]
- Romero-Meza, G.; Mugnier, M.R. Trypanosoma brucei. Trends Parasitol. 2020, 36, 571–572. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.E.; Englund, P.T. Network news: The replication of kinetoplast DNA. Annu. Rev. Microbiol. 2012, 66, 473–491. [Google Scholar] [CrossRef]
- Read, L.K.; Lukes, J.; Hashimi, H. Trypanosome RNA editing: The complexity of getting U in and taking U out. Wiley Interdiscip. Rev. RNA 2016, 7, 33–51. [Google Scholar] [CrossRef]
- Simpson, A.G.B.; Stevens, J.R.; Lukeš, J. The evolution and diversity of kinetoplastid flagellates. Trends Parasitol. 2006, 22, 168–174. [Google Scholar] [CrossRef]
- Marcili, A.; Sperança, M.A.; da Costa, A.P.; Madeira, M.d.F.; Soares, H.S.; Sanches, C.d.O.C.C.; Acosta, I.d.C.L.; Girotto, A.; Minervino, A.H.H.; Horta, M.C.; et al. Phylogenetic relationships of Leishmania species based on trypanosomatid barcode (SSU rDNA) and gGAPDH genes: Taxonomic revision of Leishmania (L.) infantum chagasi in South America. Infect. Genet. Evol. 2014, 25, 44–51. [Google Scholar] [CrossRef]
- Hamilton, P.B.; Stevens, J.R.; Gaunt, M.W.; Gidley, J.; Gibson, W.C. Trypanosomes are monophyletic: Evidence from genes for glyceraldehyde phosphate dehydrogenase and small subunit ribosomal RNA. Int. J. Parasitol. 2004, 34, 1393–1404. [Google Scholar] [CrossRef]
- Van der Auwera, G.; Dujardin, J.C. Species typing in dermal leishmaniasis. Clin. Microbiol. Rev. 2015, 28, 265–294. [Google Scholar] [CrossRef]
- Espada, C.R.; Ferreira, B.A.; Ortiz, P.A.; Uliana, S.R.B.; Coelho, A.C. Full nucleotide sequencing of ribosomal DNA internal transcribed spacer of Leishmania species causing cutaneous leishmaniasis in Brazil and its potential for species typing. Acta Trop. 2021, 223, 106093. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.; Ravel, C.; Verweij, J.J.; Bart, A.; Schönian, G.; Felger, I. Evaluation of four single-locus markers for Leishmania species discrimination by sequencing. J. Clin. Microbiol. 2014, 52, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Schönian, G.; Mauricio, I.; Cupolillo, E. Is it time to revise the nomenclature of Leishmania? Trends Parasitol. 2010, 26, 466–469. [Google Scholar] [CrossRef]
- Boité, M.C.; Mauricio, I.L.; Miles, M.A.; Cupolillo, E. New insights on taxonomy, phylogeny and population genetics of Leishmania (Viannia) parasites based on multilocus sequence analysis. PLoS Negl. Trop. Dis. 2012, 6, e1888. [Google Scholar] [CrossRef] [PubMed]
- El Baidouri, F.; Diancourt, L.; Berry, V.; Chevenet, F.; Pratlong, F.; Marty, P.; Ravel, C. Genetic structure and evolution of the Leishmania genus in Africa and Eurasia: What does MLSA tell us. PLoS Negl. Trop. Dis. 2013, 7, e2255. [Google Scholar] [CrossRef]
- Akhoundi, M.; Downing, T.; Votýpka, J.; Kuhls, K.; Lukeš, J.; Cannet, A.; Ravel, C.; Marty, P.; Delaunay, P.; Kasbari, M.; et al. Leishmania infections: Molecular targets and diagnosis. Mol. Asp. Med. 2017, 57, 1–29. [Google Scholar] [CrossRef]
- Schönian, G.; Kuhls, K.; Mauricio, I.L. Molecular approaches for a better understanding of the epidemiology and population genetics of Leishmania. Parasitology 2011, 138, 405–425. [Google Scholar] [CrossRef]
- Kaufer, A.; Stark, D.; Ellis, J. A review of the systematics, species identification and diagnostics of the Trypanosomatidae using the maxicircle kinetoplast DNA: From past to present. Int. J. Parasitol. 2020, 50, 449–460. [Google Scholar] [CrossRef]
- Camacho, E.; Rastrojo, A.; Sanchiz, Á.; González-de la Fuente, S.; Aguado, B.; Requena, J.M. Leishmania Mitochondrial Genomes: Maxicircle Structure and Heterogeneity of Minicircles. Genes 2019, 10, 758. [Google Scholar] [CrossRef]
- Akopyants, N.S.; Kimblin, N.; Secundino, N.; Patrick, R.; Peters, N.; Lawyer, P.; Dobson, D.E.; Beverley, S.M.; Sacks, D.L. Demonstration of genetic exchange during cyclical development of Leishmania in the sand fly vector. Science 2009, 324, 265–268. [Google Scholar] [CrossRef]
- Rogers, M.B.; Downing, T.; Smith, B.A.; Imamura, H.; Sanders, M.; Svobodova, M.; Volf, P.; Berriman, M.; Cotton, J.A.; Smith, D.F. Genomic confirmation of hybridisation and recent inbreeding in a vector-isolated leishmania population. PLoS Genet. 2014, 10, e1004092. [Google Scholar] [CrossRef] [PubMed]
- Callejas-Hernández, F.; Herreros-Cabello, A.; Del Moral-Salmoral, J.; Fresno, M.; Gironès, N. The Complete Mitochondrial DNA of Trypanosoma cruzi: Maxicircles and Minicircles. Front Cell Infect. Microbiol. 2021, 11, 672448. [Google Scholar] [CrossRef] [PubMed]
- Kaufer, A.; Barratt, J.; Stark, D.; Ellis, J. The complete coding region of the maxicircle as a superior phylogenetic marker for exploring evolutionary relationships between members of the Leishmaniinae. Infect. Genet. Evol. 2019, 70, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Gerasimov, E.S.; Zamyatnina, K.A.; Matveeva, N.S.; Rudenskaya, Y.A.; Kraeva, N.; Kolesnikov, A.A.; Yurchenko, V. Common Structural Patterns in the Maxicircle Divergent Region of Trypanosomatidae. Pathogens 2020, 9, 100. [Google Scholar] [CrossRef]
- Kaufer, A.; Stark, D.; Ellis, J. Evolutionary Insight into the Trypanosomatidae Using Alignment-Free Phylogenomics of the Kinetoplast. Pathogens 2019, 8, 157. [Google Scholar] [CrossRef]
- Rogers, M.B.; Hilley, J.D.; Dickens, N.J.; Wilkes, J.; Bates, P.A.; Depledge, D.P.; Harris, D.; Her, Y.; Herzyk, P.; Imamura, H.; et al. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011, 21, 2129–2142. [Google Scholar] [CrossRef]
- Real, F.; Vidal, R.O.; Carazzolle, M.F.; Mondego, J.M.; Costa, G.G.; Herai, R.H.; Würtele, M.; de Carvalho, L.M.; Carmona e Ferreira, R.; Mortara, R.A.; et al. The genome sequence of Leishmania (Leishmania) amazonensis: Functional annotation and extended analysis of gene models. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2013, 20, 567–581. [Google Scholar] [CrossRef]
- Llanes, A.; Restrepo, C.M.; Del Vecchio, G.; Anguizola, F.J.; Lleonart, R. The genome of Leishmania panamensis: Insights into genomics of the L. (Viannia) subgenus. Sci. Rep. 2015, 5, 8550. [Google Scholar] [CrossRef]
- Teixeira, D.G.; Monteiro, G.R.G.; Martins, D.R.A.; Fernandes, M.Z.; Macedo-Silva, V.; Ansaldi, M.; Nascimento, P.R.P.; Kurtz, M.A.; Streit, J.A.; Ximenes, M.; et al. Comparative analyses of whole genome sequences of Leishmania infantum isolates from humans and dogs in northeastern Brazil. Int. J. Parasitol. 2017, 47, 655–665. [Google Scholar] [CrossRef]
- Valdivia, H.O.; Almeida, L.V.; Roatt, B.M.; Reis-Cunha, J.L.; Pereira, A.A.S.; Gontijo, C.; Fujiwara, R.T.; Reis, A.B.; Sanders, M.J.; Cotton, J.A.; et al. Comparative genomics of canine-isolated Leishmania (Leishmania) amazonensis from an endemic focus of visceral leishmaniasis in Governador Valadares, southeastern Brazil. Sci. Rep. 2017, 7, 40804. [Google Scholar] [CrossRef]
- Gonzalez-de la Fuente, S.; Peiro-Pastor, R.; Rastrojo, A.; Moreno, J.; Carrasco-Ramiro, F.; Requena, J.M.; Aguado, B. Resequencing of the Leishmania infantum (strain JPCM5) genome and de novo assembly into 36 contigs. Sci. Rep. 2017, 7, 18050. [Google Scholar] [CrossRef] [PubMed]
- Camacho, E.; González-de la Fuente, S.; Rastrojo, A.; Peiró-Pastor, R.; Solana, J.C.; Tabera, L.; Gamarro, F.; Carrasco-Ramiro, F.; Requena, J.M.; Aguado, B. Complete assembly of the Leishmania donovani (HU3 strain) genome and transcriptome annotation. Sci. Rep. 2019, 9, 6127. [Google Scholar] [CrossRef] [PubMed]
- Camacho, E.; González-de la Fuente, S.G.-d.l.; Solana, J.C.; Rastrojo, A.; Carrasco-Ramiro, F.; Requena, J.M.; Aguado, B. Gene Annotation and Transcriptome Delineation on a De Novo Genome Assembly for the Reference Leishmania major Friedlin Strain. Genes 2021, 12, 1359. [Google Scholar] [CrossRef] [PubMed]
- Aslett, M.; Aurrecoechea, C.; Berriman, M.; Brestelli, J.; Brunk, B.P.; Carrington, M.; Depledge, D.P.; Fischer, S.; Gajria, B.; Gao, X.; et al. TriTrypDB: A functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010, 38, D457–D462. [Google Scholar] [CrossRef]
- Bussotti, G.; Benkahla, A.; Jeddi, F.; Souiaï, O.; Aoun, K.; Späth, G.F.; Bouratbine, A. Nuclear and mitochondrial genome sequencing of North-African Leishmania infantum isolates from cured and relapsed visceral leishmaniasis patients reveals variations correlating with geography and phenotype. Microb. Genom. 2020, 6, e000444. [Google Scholar] [CrossRef]
- De la Cruz, V.F.; Neckelmann, N.; Simpson, L. Sequences of six genes and several open reading frames in the kinetoplast maxicircle DNA of Leishmania tarentolae. J. Biol. Chem. 1984, 259, 15136–15147. [Google Scholar] [CrossRef]
- Urrea, D.A.; Triana-Chavez, O.; Alzate, J.F. Mitochondrial genomics of human pathogenic parasite Leishmania (Viannia) panamensis. PeerJ 2019, 7, e7235. [Google Scholar] [CrossRef]
- Greif, G.; Rodriguez, M.; Reyna-Bello, A.; Robello, C.; Alvarez-Valin, F. Kinetoplast adaptations in American strains from Trypanosoma vivax. Mutat. Res. 2015, 773, 69–82. [Google Scholar] [CrossRef]
- Sloof, P.; de Haan, A.; Eier, W.; van Iersel, M.; Boel, E.; van Steeg, H.; Benne, R. The nucleotide sequence of the variable region in Trypanosoma brucei completes the sequence analysis of the maxicircle component of mitochondrial kinetoplast DNA. Mol. Biochem. Parasitol. 1992, 56, 289–299. [Google Scholar] [CrossRef]
- Botero, A.; Kapeller, I.; Cooper, C.; Clode, P.L.; Shlomai, J.; Thompson, R.C.A. The kinetoplast DNA of the Australian trypanosome, Trypanosoma copemani, shares features with Trypanosoma cruzi and Trypanosoma lewisi. Int. J. Parasitol. 2018, 48, 691–700. [Google Scholar] [CrossRef]
- Lin, R.H.; Lai, D.H.; Zheng, L.L.; Wu, J.; Lukes, J.; Hide, G.; Lun, Z.R. Analysis of the mitochondrial maxicircle of Trypanosoma lewisi, a neglected human pathogen. Parasites Vectors 2015, 8, 665. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Westenberger, S.J.; Cerqueira, G.C.; El-Sayed, N.M.; Zingales, B.; Campbell, D.A.; Sturm, N.R. Trypanosoma cruzi mitochondrial maxicircles display species- and strain-specific variation and a conserved element in the non-coding region. BMC Genom. 2006, 7, 60. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence Limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L. The Mitochondrial Genome of Kinetoplastid Protozoa: Genomic Organization, Transcription, Replication, and Evolution. Annu. Rev. Microbiol. 1987, 41, 363–380. [Google Scholar] [CrossRef]
- Simpson, L.; Douglass, S.M.; Lake, J.A.; Pellegrini, M.; Li, F. Comparison of the Mitochondrial Genomes and Steady State Transcriptomes of Two Strains of the Trypanosomatid Parasite, Leishmania tarentolae. PLoS Negl. Trop. Dis. 2015, 9, e0003841. [Google Scholar] [CrossRef]
- Flegontov, P.N.; Guo, Q.; Ren, L.; Strelkova, M.V.; Kolesnikov, A.A. Conserved repeats in the kinetoplast maxicircle divergent region of Leishmania sp. and Leptomonas seymouri. Mol. Genet. Genom. MGG 2006, 276, 322–333. [Google Scholar] [CrossRef]
- Simpson, L.; Shaw, J. RNA editing and the mitochondrial cryptogenes of kinetoplastid protozoa. Cell 1989, 57, 355–366. [Google Scholar] [CrossRef]
- Asato, Y.; Oshiro, M.; Myint, C.K.; Yamamoto, Y.; Kato, H.; Marco, J.D.; Mimori, T.; Gomez, E.A.; Hashiguchi, Y.; Uezato, H. Phylogenic analysis of the genus Leishmania by cytochrome b gene sequencing. Exp. Parasitol. 2009, 121, 352–361. [Google Scholar] [CrossRef]
- Kuhls, K.; Mauricio, I. Phylogenetic Studies. Methods Mol. Biol. 2019, 1971, 9–68. [Google Scholar] [CrossRef] [PubMed]
- Lukeš, J.; Skalický, T.; Týč, J.; Votýpka, J.; Yurchenko, V. Evolution of parasitism in kinetoplastid flagellates. Mol. Biochem. Parasitol. 2014, 195, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Barratt, J.; Kaufer, A.; Peters, B.; Craig, D.; Lawrence, A.; Roberts, T.; Lee, R.; McAuliffe, G.; Stark, D.; Ellis, J. Isolation of Novel Trypanosomatid, Zelonia australiensis sp. nov. (Kinetoplastida: Trypanosomatidae) Provides Support for a Gondwanan Origin of Dixenous Parasitism in the Leishmaniinae. PLoS Negl. Trop. Dis. 2017, 11, e0005215. [Google Scholar] [CrossRef] [PubMed]
- Fraga, J.; Fernández-Calienes, A.; Montalvo, A.M.; Maes, I.; Deborggraeve, S.; Büscher, P.; Dujardin, J.C.; Van der Auwera, G. Phylogenetic analysis of the Trypanosoma genus based on the heat-shock protein 70 gene. Infect. Genet. Evol. 2016, 43, 165–172. [Google Scholar] [CrossRef]
- Van den Broeck, F.; Savill, N.J.; Imamura, H.; Sanders, M.; Maes, I.; Cooper, S.; Mateus, D.; Jara, M.; Adaui, V.; Arevalo, J.; et al. Ecological divergence and hybridization of Neotropical Leishmania parasites. Proc. Natl. Acad. Sci. USA 2020, 117, 25159–25168. [Google Scholar] [CrossRef]
- Akhoundi, M.; Kuhls, K.; Cannet, A.; Votypka, J.; Marty, P.; Delaunay, P.; Sereno, D. A Historical Overview of the Classification, Evolution, and Dispersion of Leishmania Parasites and Sandflies. PLoS Negl. Trop. Dis. 2016, 10, e0004349. [Google Scholar] [CrossRef]
- Paranaiba, L.F.; Pinheiro, L.J.; Torrecilhas, A.C.; Macedo, D.H.; Menezes-Neto, A.; Tafuri, W.L.; Soares, R.P. Leishmania enriettii (Muniz & Medina, 1948): A highly diverse parasite is here to stay. PLoS Pathog. 2017, 13, e1006303. [Google Scholar] [CrossRef]
- Fraga, J.; Montalvo, A.M.; De Doncker, S.; Dujardin, J.C.; Van der Auwera, G. Phylogeny of Leishmania species based on the heat-shock protein 70 gene. Infect. Genet. Evol. 2010, 10, 238–245. [Google Scholar] [CrossRef]
- Franssen, S.U.; Durrant, C.; Stark, O.; Moser, B.; Downing, T.; Imamura, H.; Dujardin, J.C.; Sanders, M.J.; Mauricio, I.; Miles, M.A.; et al. Global genome diversity of the Leishmania donovani complex. eLife 2020, 9, e51243. [Google Scholar] [CrossRef]
- Grace, C.A.; Forrester, S.; Silva, V.C.; Carvalho, K.S.S.; Kilford, H.; Chew, Y.P.; James, S.; Costa, D.L.; Mottram, J.C.; Costa, C.; et al. Candidates for Balancing Selection in Leishmania donovani Complex Parasites. Genome Biol. Evol. 2021, 13, evab265. [Google Scholar] [CrossRef]
- Carvalho, K.S.S.; da Silva Júnior, W.J.; da Silveira Regueira Neto, M.; Silva, V.C.; de Sá Leitão Paiva Júnior, S.; Balbino, V.Q.; Costa, D.L.; Costa, C.H.N. Application of next generation sequencing (NGS) for descriptive analysis of 30 genomes of Leishmania infantum isolates in Middle-North Brazil. Sci. Rep. 2020, 10, 12321. [Google Scholar] [CrossRef] [PubMed]
- Requena, J.M.; Solana, J.C. Maxicircles_Linf-Carvalho_2020. Mendeley Data, V1. 2022. Available online: https://doi.org/10.17632/rmjc4wmr4x.1 (accessed on 12 June 2022).
- Bussotti, G.; Gouzelou, E.; Cortes Boite, M.; Kherachi, I.; Harrat, Z.; Eddaikra, N.; Mottram, J.C.; Antoniou, M.; Christodoulou, V.; Bali, A.; et al. Leishmania Genome Dynamics during Environmental Adaptation Reveal Strain-Specific Differences in Gene Copy Number Variation, Karyotype Instability, and Telomeric Amplification. MBio 2018, 9, e01399-18. [Google Scholar] [CrossRef] [PubMed]
- Requena, J.M.; Solana, J.C. Maxicircles_Linf-Bussotti_2018. Mendeley Data, V1. 2022. Available online: https://doi.org/10.17632/twkkb6bmdv.1 (accessed on 12 June 2022).
- Requena, J.M.; Solana, J.C. Maxicircles_Rogers_2014. Mendeley Data, V1. 2022. Available online: https://doi.org/10.17632/vh55y46yr4.1 (accessed on 12 June 2022).
- Requena, J.M.; Solana, J.C. Maxicircles_Linf-Franssen_2020. Mendeley Data, V1. 2022. Available online: https://doi.org/10.17632/nnf649r9kf.1 (accessed on 12 June 2022).
- Requena, J.M.; Solana, J.C. Maxicircles_Linf-Bussotti_2020. Mendeley Data, V1. 2022. Available online: https://doi.org/10.17632/nwm8dw97sg.1 (accessed on 12 June 2022).
- Requena, J.M.; Solana, J.C. Maxicircles_Linf-Solana_2022. Mendeley Data, V1. 2022. Available online: https://doi.org/10.17632/w65x97kng9.1 (accessed on 12 June 2022).
- Leblois, R.; Kuhls, K.; François, O.; Schönian, G.; Wirth, T. Guns, germs and dogs: On the origin of Leishmania chagasi. Infect. Genet. Evol. 2011, 11, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Svobodová, M.; Alten, B.; Zídková, L.; Dvorák, V.; Hlavacková, J.; Mysková, J.; Seblová, V.; Kasap, O.E.; Belen, A.; Votýpka, J.; et al. Cutaneous leishmaniasis caused by Leishmania infantum transmitted by Phlebotomus tobbi. Int. J. Parasitol. 2009, 39, 251–256. [Google Scholar] [CrossRef]
- Gouzelou, E.; Haralambous, C.; Amro, A.; Mentis, A.; Pratlong, F.; Dedet, J.-P.; Votypka, J.; Volf, P.; Ozensoy Toz, S.; Kuhls, K.; et al. Multilocus Microsatellite Typing (MLMT) of Strains from Turkey and Cyprus Reveals a Novel Monophyletic L. donovani Sensu Lato Group. PLoS Negl. Trop. Dis. 2012, 6, e1507. [Google Scholar] [CrossRef]
- Chicharro, C.; Llanes-Acevedo, I.P.; Garcia, E.; Nieto, J.; Moreno, J.; Cruz, I. Molecular typing of Leishmania infantum isolates from a leishmaniasis outbreak in Madrid, Spain, 2009 to 2012. Eurosurveillance 2013, 18, 20545. [Google Scholar] [CrossRef]
- Martin-Martin, I.; Jimenez, M.; Gonzalez, E.; Eguiluz, C.; Molina, R. Natural transmission of Leishmania infantum through experimentally infected Phlebotomus perniciosus highlights the virulence of Leishmania parasites circulating in the human visceral leishmaniasis outbreak in Madrid, Spain. Vet. Res. 2015, 46, 138. [Google Scholar] [CrossRef]
- Mas, A.; Martínez-Rodrigo, A.; Orden, J.A.; Molina, R.; Jiménez, M.; Jiménez, M.; Carrión, J.; Domínguez-Bernal, G. Properties of virulence emergence of Leishmania infantum isolates from Phlebotomus perniciosus collected during the human leishmaniosis outbreak in Madrid, Spain. Hepatic histopathology and immunological parameters as virulence markers in the mouse model. Transbound Emerg. Dis. 2021, 68, 704–714. [Google Scholar] [CrossRef]
- Chargui, N.; Haouas, N.; Slama, D.; Gorcii, M.; Jaouadi, K.; Essabbah-Aguir, N.; Mezhoud, H.; Babba, H. Transmission of visceral leishmaniasis in a previously non-endemic region of Tunisia: Detection of Leishmania DNA in Phlebotomus perniciosus. J. Vector Ecol. 2013, 38, 1–5. [Google Scholar] [CrossRef]
- Ravel, C.; Cortes, S.; Pratlong, F.; Morio, F.; Dedet, J.P.; Campino, L. First report of genetic hybrids between two very divergent Leishmania species: Leishmania infantum and Leishmania major. Int. J. Parasitol. 2006, 36, 1383–1388. [Google Scholar] [CrossRef]
- Kato, H.; Cáceres, A.G.; Gomez, E.A.; Tabbabi, A.; Mizushima, D.; Yamamoto, D.S.; Hashiguchi, Y. Prevalence of Genetically Complex Leishmania Strains with Hybrid and Mito-Nuclear Discordance. Front. Cell. Infect. Microbiol. 2021, 11, 625001. [Google Scholar] [CrossRef] [PubMed]


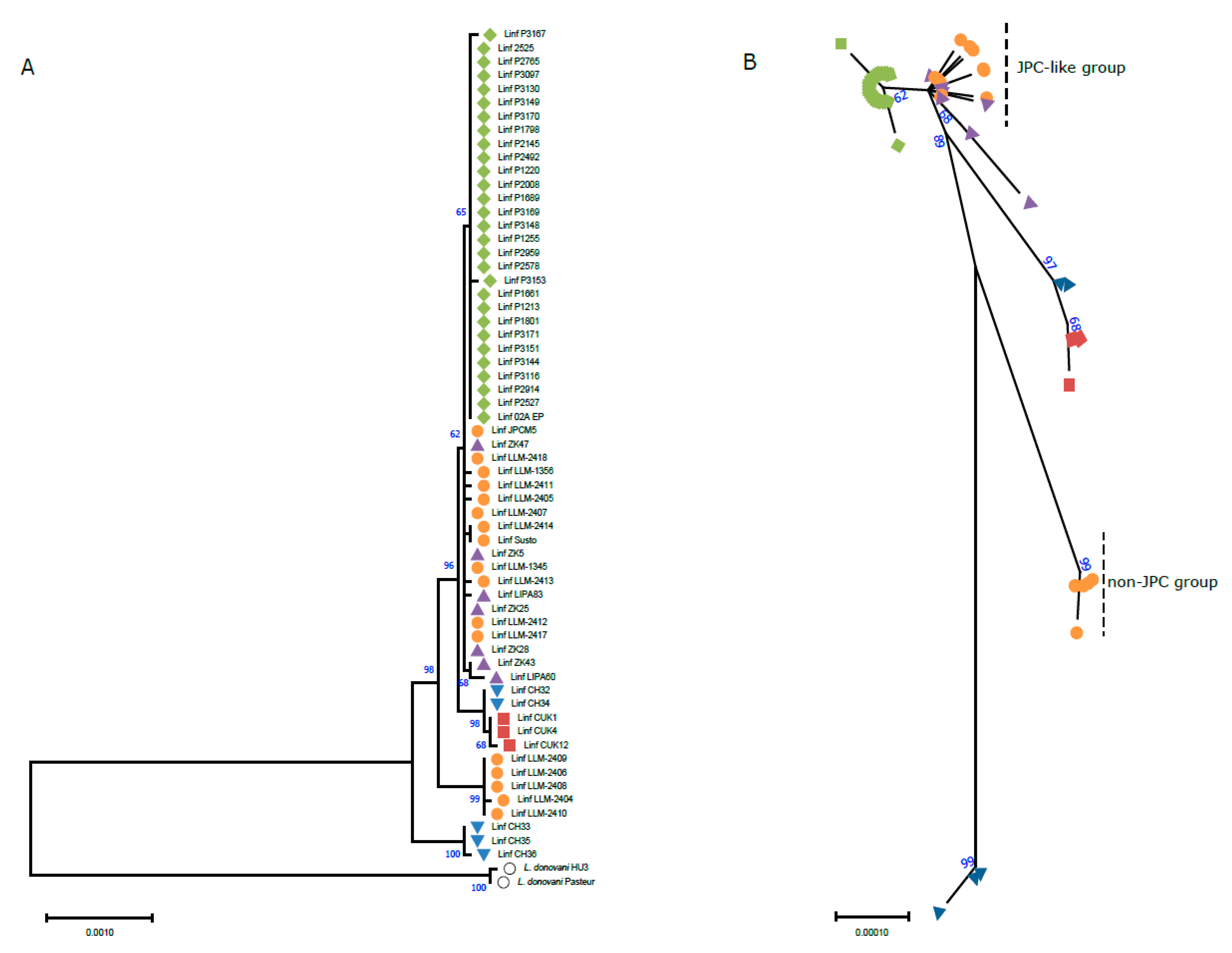
 Brazilian isolates;
Brazilian isolates;  Spanish isolates;
Spanish isolates;  Tunisian isolates;
Tunisian isolates;  Turkish isolates;
Turkish isolates;  Cypriot isolates;
Cypriot isolates;  L. donovani (outgroup). All the evolutionary analyses were conducted in MEGA 11 [45].
Brazilian isolates; Spanish isolates; Tunisian isolates; Turkish isolates; Cypriot isolates; L. donovani (outgroup). All the evolutionary analyses were conducted in MEGA 11 [45].
L. donovani (outgroup). All the evolutionary analyses were conducted in MEGA 11 [45].
Brazilian isolates; Spanish isolates; Tunisian isolates; Turkish isolates; Cypriot isolates; L. donovani (outgroup). All the evolutionary analyses were conducted in MEGA 11 [45].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | GenBank/ENA * | Reference | Mendeley Data (DOI) a |
|---|---|---|---|
| Leishmania aethiopica L147 MHOM/ET/1972/L100 | SRR834802 | This work | 10.17632/8h8tzrbzft.1 |
| Leishmania tropica L590 MHOM/IL/1990/P283 | SRR834907 | This work | 10.17632/23t2h58cvr.1 |
| Leishmania major MHOM/IL/81/Friedlin | LR697138 | [19] | 10.17632/hdyj8hbt39.1 |
| L. infantum JPCM5 MCAN/ES/98/LLM-724 | LR697137 | [19] | 10.17632/nszm7rb8y7.1 |
| Leishmania donovani Pasteur | CP022652.1 | Direct submission | 10.17632/jhy362m5ms.1 |
| Leishmania donovani HU3 MHOM/ET/67/HU3 | ERR2191875 | This work | 10.17632/74d3b3pnvt.1 |
| Leishmania amazonensis IFLA/BR/1967/PH8 | SRR8584809 | This work | 10.17632/pfdfzpwgjd.1 |
| Leishmania adleri MARV/ET/75/HO174 | LR697136 | [19] | 10.17632/98cn9h5t67.1 |
| Leishmania tarentolae | M10126.1 | [36] | 10.17632/6kwj82nt8s.1 |
| Leishmania guyanensis LgCL085 | LR697135 | [19] | 10.17632/rcjzz74fvj.1 |
| Leishmania panamensis UA946 | MK570510.1 | [37] | 10.17632/9sxnsjy9xx.1 |
| Leishmania braziliensis MHOM/BR/75/M2904 | LR697134 | [19] | 10.17632/bg3 × 4tcr64.1 |
| Leishmania peruviana MHOM/PE/90/LCA0482 | ERR3656053 | This work | 10.17632/4wf4hb3k7g.1 |
| Leishmania lainsoni MHOM/BR/1981/M6426 | SRR1657912 | This work | 10.17632/k6ffm6j8c3.1 |
| Leishmania enrietti LV763 MCAV/BR/2001/CUR178 | SRR13558795 | This work | 10.17632/xhj67pv882.1 |
| Leishmania martiniquensis LV760 MHOM/TH/2012/LSCM1 | SRR13558784 | This work | 10.17632/9dykktyxp5.1 |
| Zelonia australiensis JB/C | MK514117.1 | [23] | 10.17632/c4zvyzftfg.1 |
| Angomonas deanei | KJ778684.1 | Direct submission | 10.17632/88w6bk3mkr.1 |
| Trypanosoma vivax Liem176 | KM386509.1 | [38] | 10.17632/484wfgchdr.1 |
| Trypanosoma vivax MT1 | KM386508.1 | [38] | 10.17632/wmkxhtz5rg.1 |
| T. brucei brucei s427 | M94286.1 | [39] | 10.17632/7yrtcn4nkk.1 |
| Trypanosoma copemani G1 | MG948557.1 | [40] | 10.17632/g44sr7djp4.1 |
| Trypanosoma lewisi CPO 02 | KR072974.1 | [41] | 10.17632/6w4gdb9rg8.1 |
| Trypanosoma rangeli SC58 | KJ803830.1 | Direct submission | 10.17632/gs6sbtbh7z.1 |
| T. cruzi Esmeraldo | DQ343646.1 | [42] | 10.17632/7hfpy5frmv.1 |
| T. cruzi CL Brener | DQ343645.1 | [42] | 10.17632/7gtbgwjjv8.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solana, J.C.; Chicharro, C.; García, E.; Aguado, B.; Moreno, J.; Requena, J.M. Assembly of a Large Collection of Maxicircle Sequences and Their Usefulness for Leishmania Taxonomy and Strain Typing. Genes 2022, 13, 1070. https://doi.org/10.3390/genes13061070
Solana JC, Chicharro C, García E, Aguado B, Moreno J, Requena JM. Assembly of a Large Collection of Maxicircle Sequences and Their Usefulness for Leishmania Taxonomy and Strain Typing. Genes. 2022; 13(6):1070. https://doi.org/10.3390/genes13061070
Chicago/Turabian StyleSolana, Jose Carlos, Carmen Chicharro, Emilia García, Begoña Aguado, Javier Moreno, and Jose M. Requena. 2022. "Assembly of a Large Collection of Maxicircle Sequences and Their Usefulness for Leishmania Taxonomy and Strain Typing" Genes 13, no. 6: 1070. https://doi.org/10.3390/genes13061070
APA StyleSolana, J. C., Chicharro, C., García, E., Aguado, B., Moreno, J., & Requena, J. M. (2022). Assembly of a Large Collection of Maxicircle Sequences and Their Usefulness for Leishmania Taxonomy and Strain Typing. Genes, 13(6), 1070. https://doi.org/10.3390/genes13061070