
Evolutionary Divergence and Radula Diversification in Two Ecomorphs from an Adaptive Radiation of Freshwater Snails

 ,
, 
Abstract
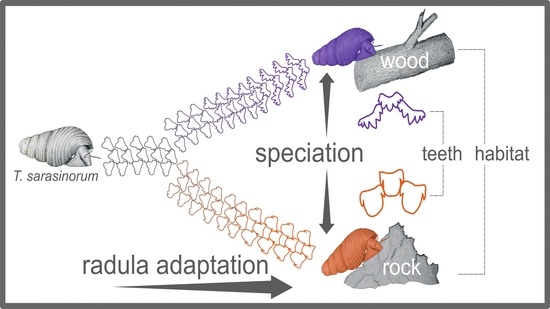
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
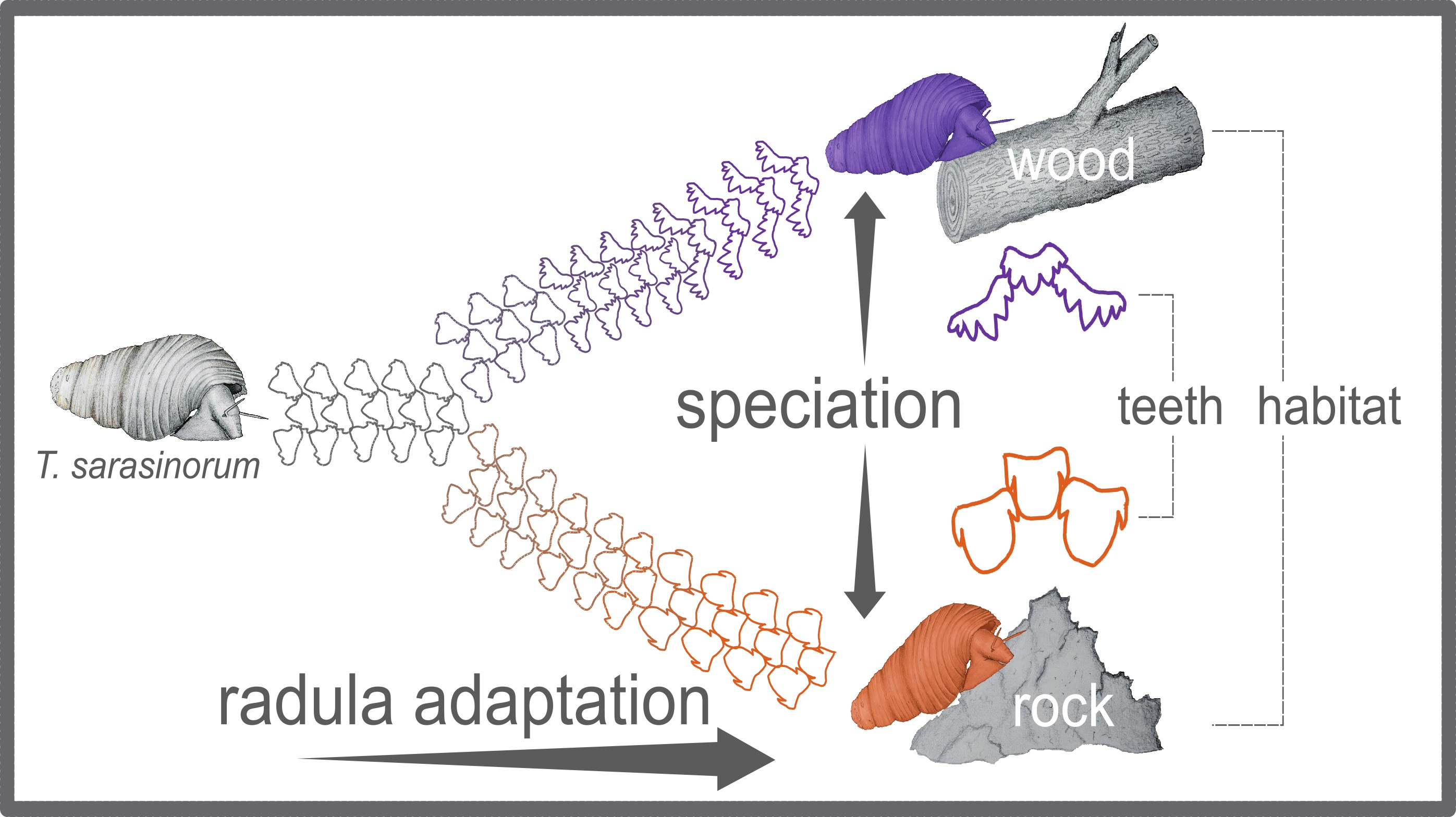
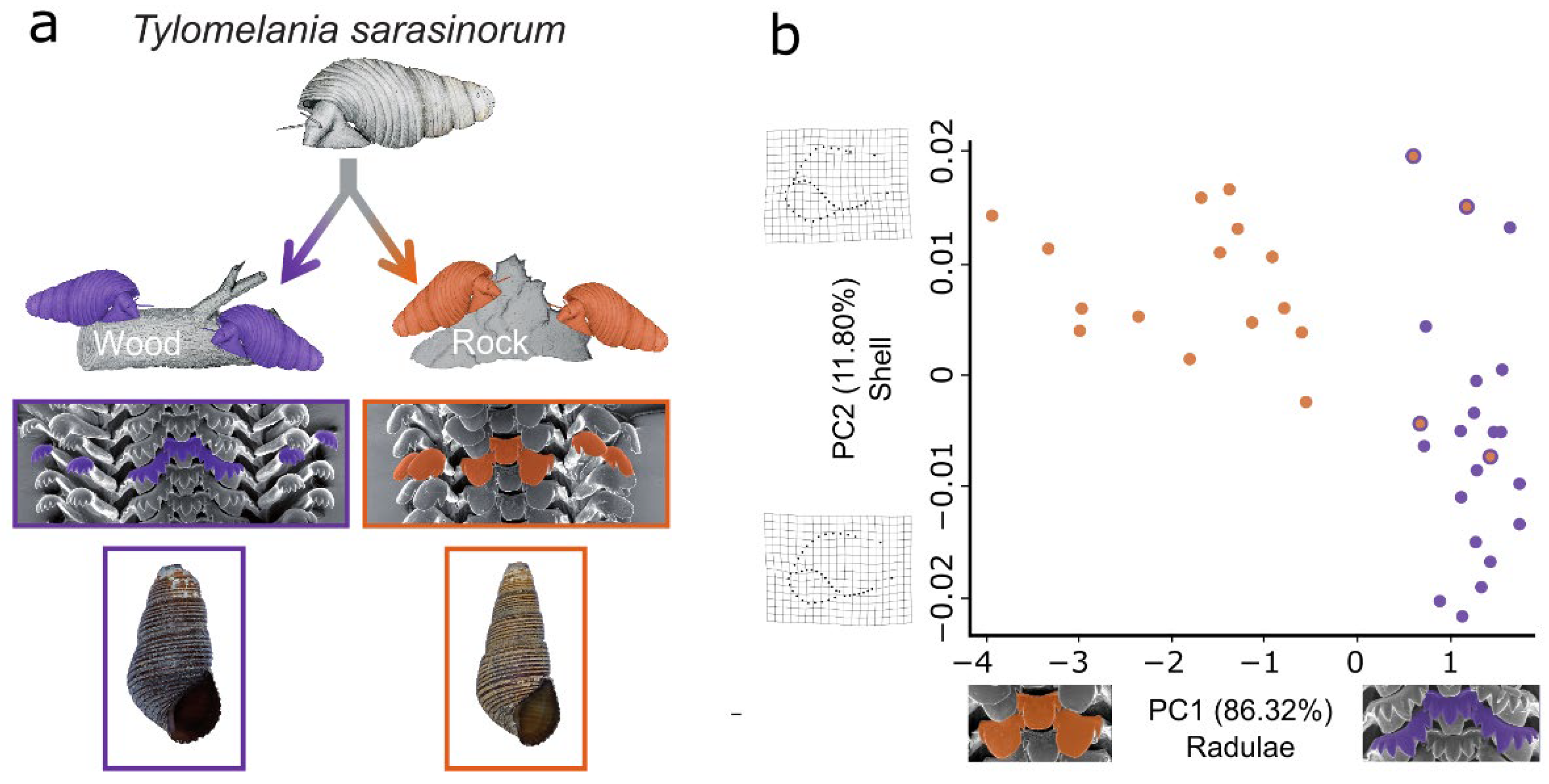
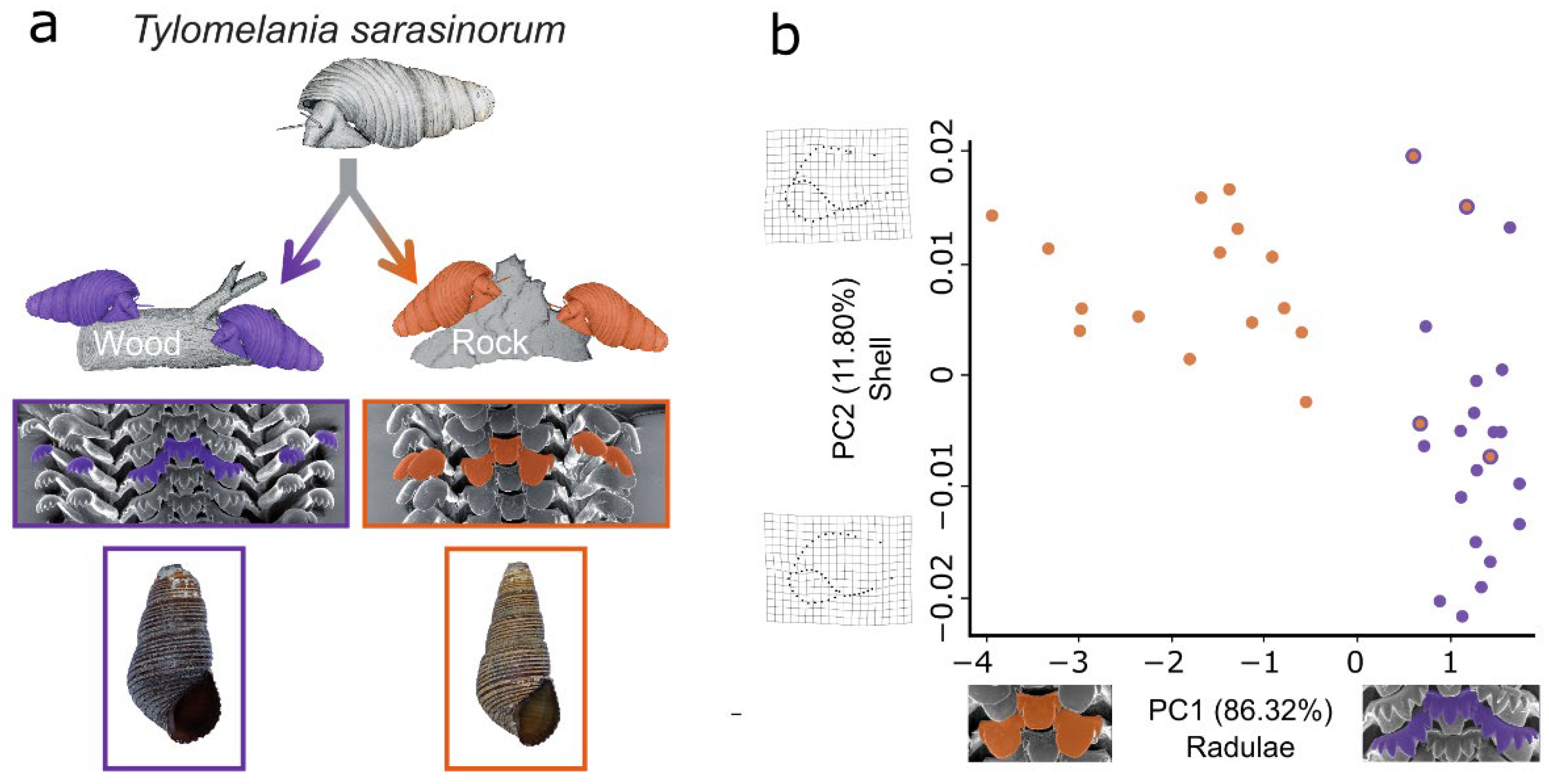
3.1. Geometric Morphometrics Corroborates a Habitat-Correlated Radula Polymorphism
3.2. Transcriptome Sequencing and Assembly
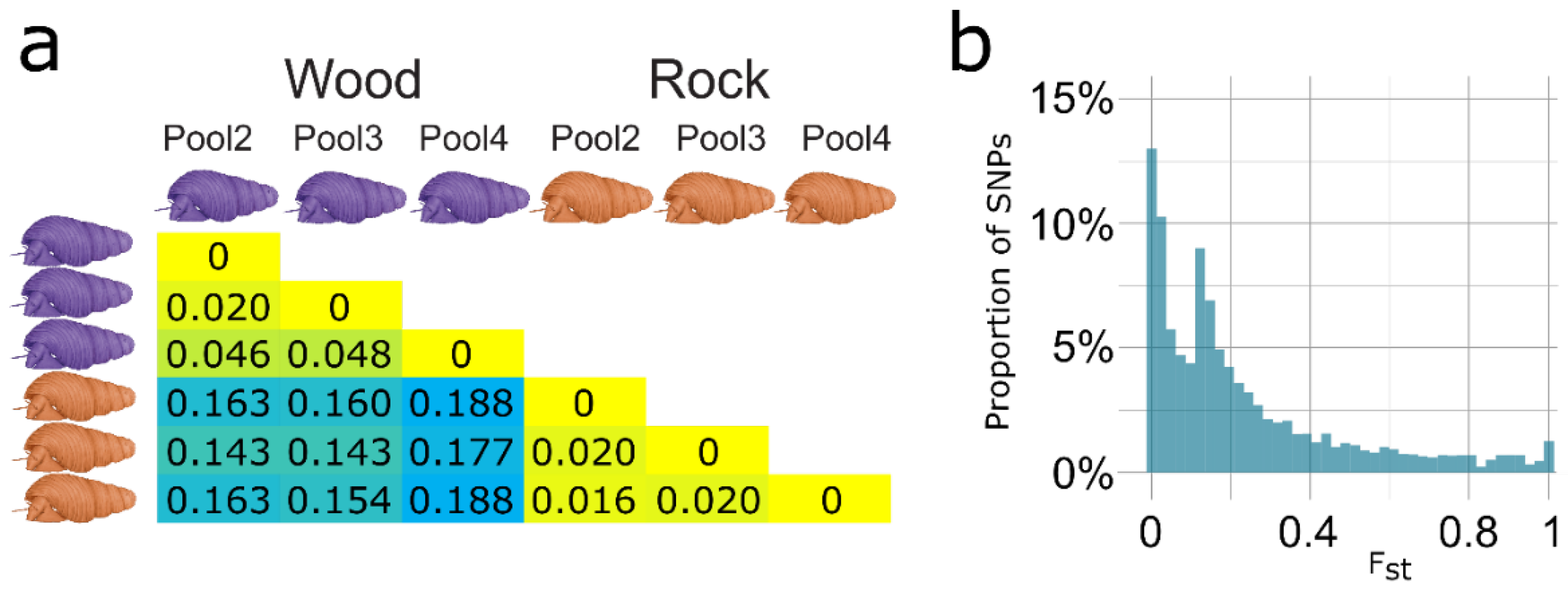
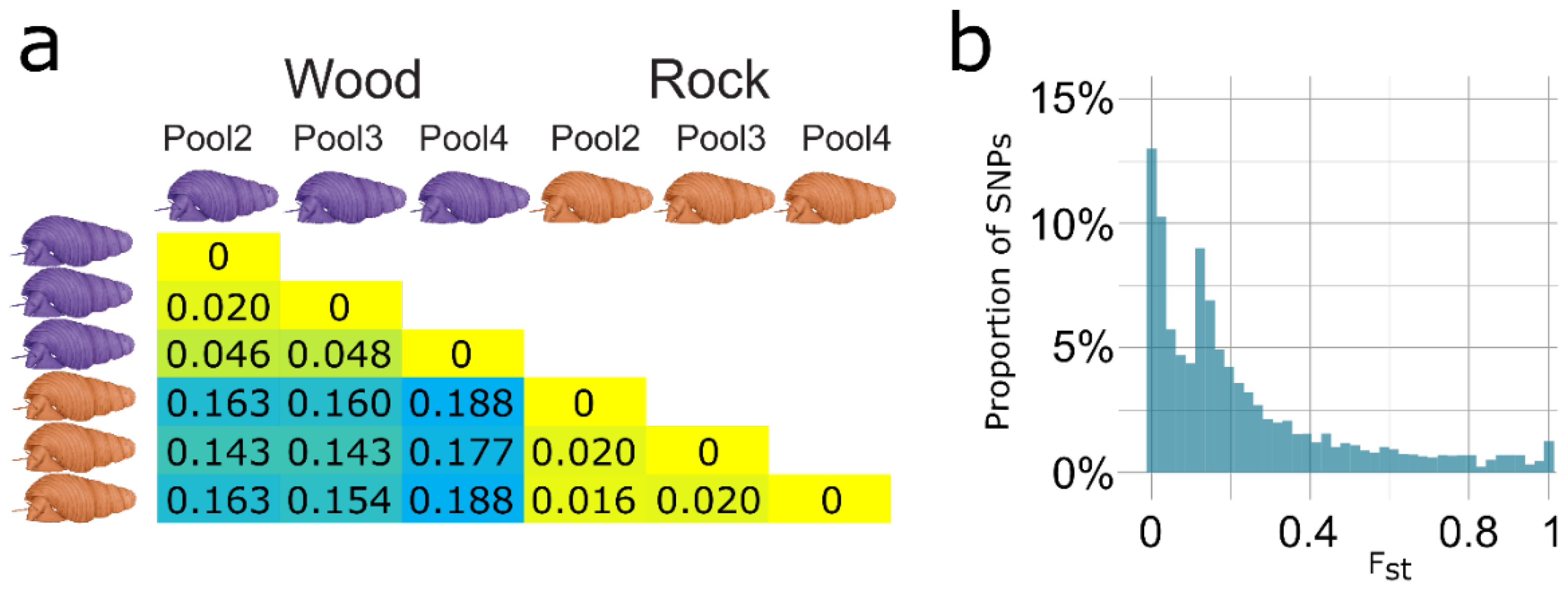
3.3. Transcriptome-Wide SNP Data Indicates Evolutionary Divergence of Ecomorphs
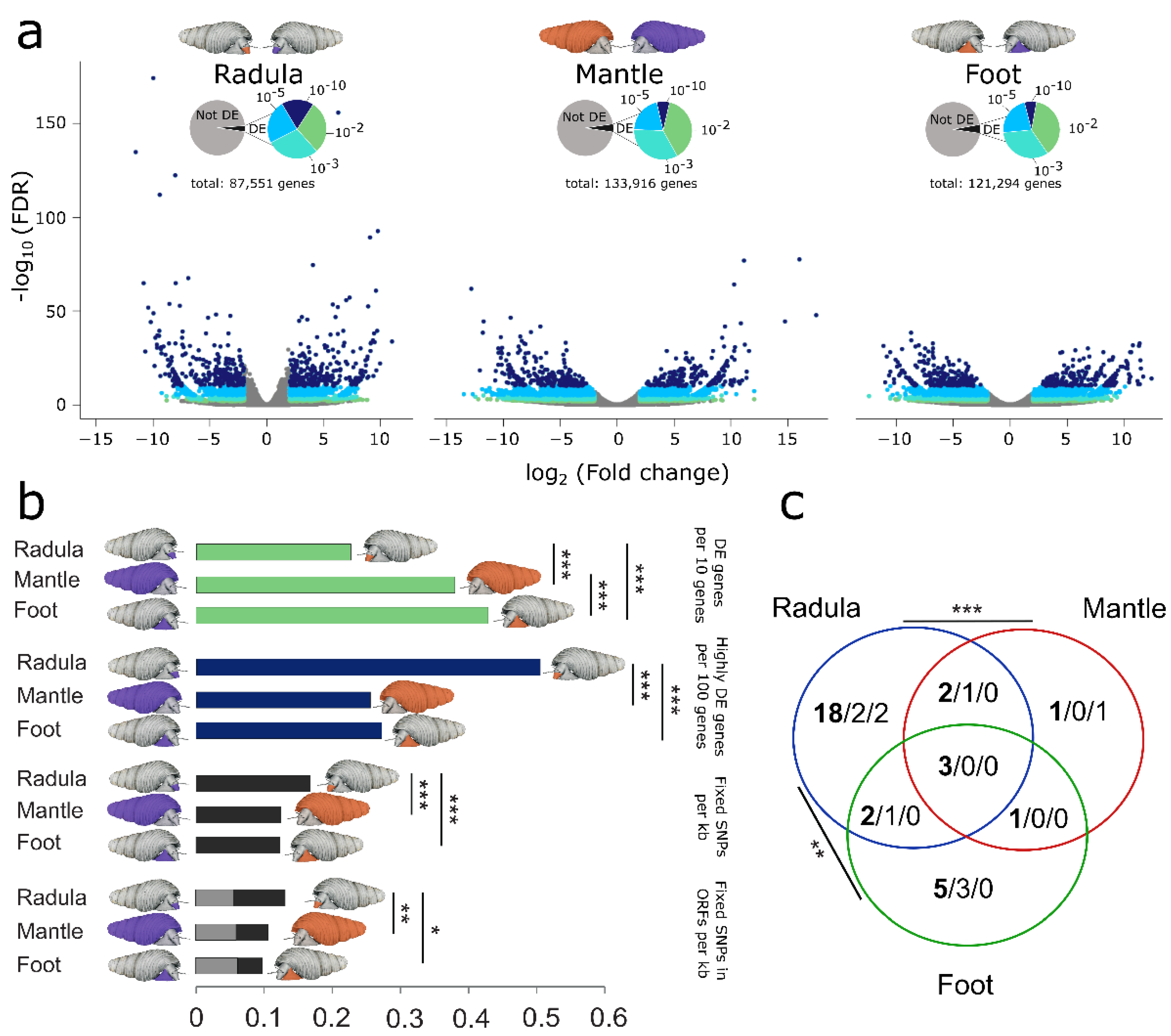
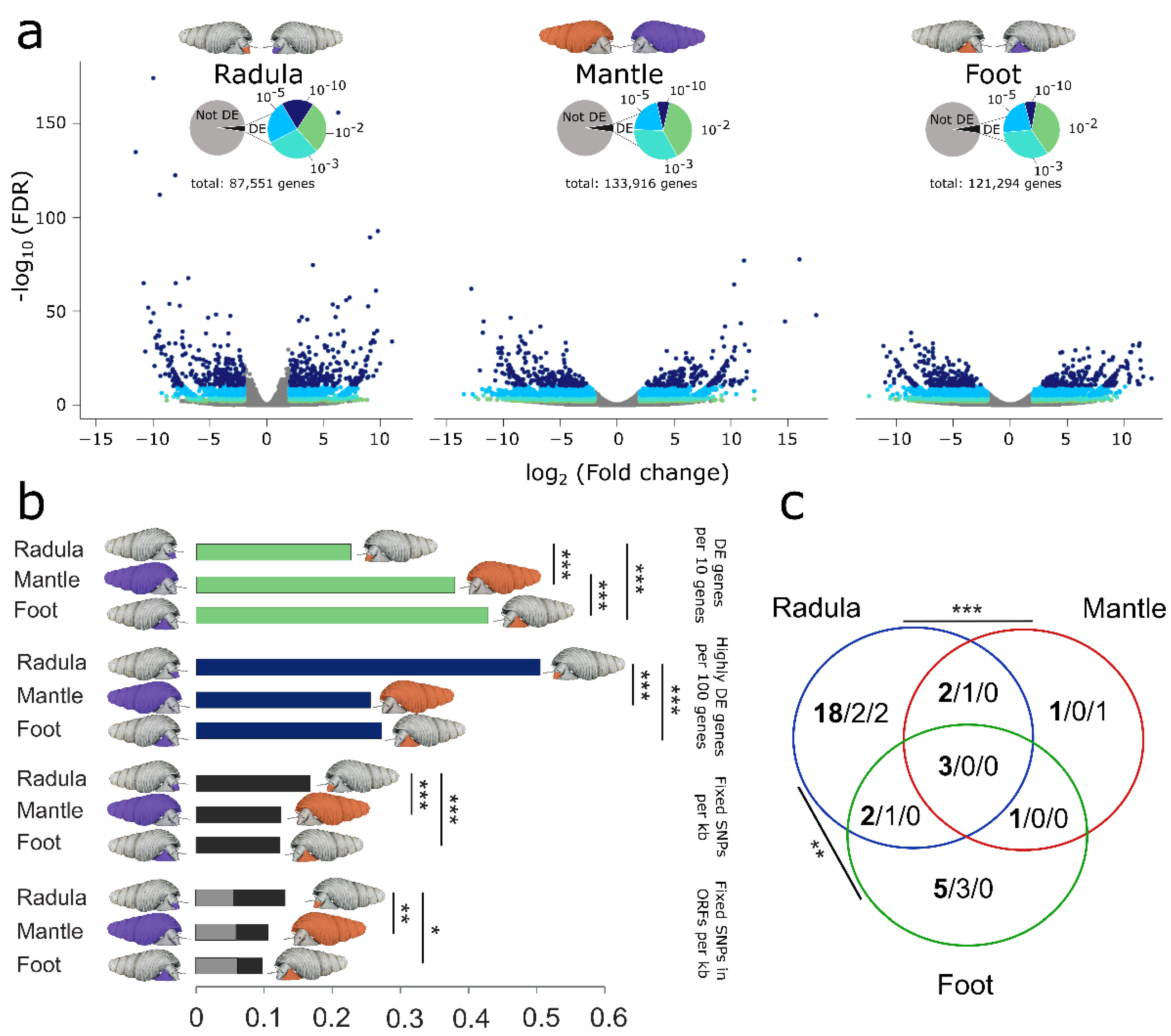
3.4. Ecomorphs Differ in Gene Expression across all Investigated Tissues
3.5. Radula Transcriptomes Exhibit Increased Sequence Divergence and Elevated Proportion of Highly DE Genes
3.6. Homologs of Candidate Genes for Radula Disparity Contributed to Craniofacial Diversification in Vertebrate Radiations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seehausen, O. African cichlid fish: A model system in adaptive radiation research. Proc. R. Soc. B Biol. Sci. 2006, 273, 1987–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berner, D.; Salzburger, W. The genomics of organismal diversification illuminated by adaptive radiations. Trends Genet. 2015, 31, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Grant, P.R.; Grant, B.R. Evolution of character displacement in Darwin’s finches. Science 2006, 313, 224–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schluter, D. The Ecology of Adaptive Radiation; Oxford University Press: Oxford, UK, 2000; ISBN 0191588326. [Google Scholar]
- Darwin, C. On the Origin of the Species by Means of Natural Selection: Or, The Preservation of Favoured Races in the Struggle for Life; John Murray: London, UK, 1859. [Google Scholar]
- Pfaender, J.; Schliewen, U.K.; Herder, F. Phenotypic traits meet patterns of resource use in the radiation of “sharpfin” sailfin silverside fish in Lake Matano. Evol. Ecol. 2010, 24, 957–974. [Google Scholar] [CrossRef]
- Elmer, K.R.; Fan, S.; Kusche, H.; Luise Spreitzer, M.; Kautt, A.F.; Franchini, P.; Meyer, A. Parallel evolution of Nicaraguan crater lake cichlid fishes via non-parallel routes. Nat. Commun. 2014, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kocher, T.D. Adaptive evolution and explosive speciation: The cichlid fish model. Nat. Rev. Genet. 2004, 5, 288–298. [Google Scholar] [CrossRef]
- Pfaender, J.; Hadiaty, R.K.; Schliewen, U.K.; Herder, F. Rugged adaptive landscapes shape a complex, sympatric radiation. Proc. R. Soc. London B 2016, 283, 20152342. [Google Scholar] [CrossRef]
- Kautt, A.F.; Kratochwil, C.F.; Nater, A.; Machado-Schiaffino, G.; Olave, M.; Henning, F.; Torres-Dowdall, J.; Härer, A.; Hulsey, C.D.; Franchini, P.; et al. Contrasting signatures of genomic divergence during sympatric speciation. Nature 2020, 588, 106–111. [Google Scholar] [CrossRef]
- Ferris, K.G.; Barnett, L.L.; Blackman, B.K.; Willis, J.H. The genetic architecture of local adaptation and reproductive isolation in sympatry within the Mimulus guttatus species complex. Mol. Ecol. 2016, 26, 208–224. [Google Scholar] [CrossRef]
- Rogers, S.; Bernatchez, L. The genetic architecture of ecological speciation and the association with signatures of selection in natural lake whitefish (Coregonus sp. Salmonidae) species pairs. Mol. Biol. Evol. 2007, 24, 1423–1438. [Google Scholar] [CrossRef]
- Salzburger, W. Understanding explosive diversification through cichlid fish genomics. Nat. Rev. Genet. 2018, 19, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Brawand, D.; Wagner, C.E.; Li, Y.I.; Malinsky, M.; Keller, I.; Fan, S.; Simakov, O.; Ng, A.Y.; Lim, Z.W.; Bezault, E.; et al. The genomic substrate for adaptive radiation in African cichlid fish. Nature 2014, 513, 375–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennenmoser, S.; Vamosi, S.M.; Nolte, A.W.; Rogers, S.M. Adaptive genomic divergence under high gene flow between freshwater and brackish-water ecotypes of prickly sculpin (Cottus asper) revealed by Pool-Seq. Mol. Ecol. 2017, 26, 25–42. [Google Scholar] [CrossRef]
- Richards, E.; Martin, C. Adaptive introgression from distant Caribbean islands contributed to the diversification of a microendemic radiation of trophic specialist pupfishes. PLoS Genet. 2017, 13, e1006919. [Google Scholar] [CrossRef] [Green Version]
- Almén, M.S.; Lamichhaney, S.; Berglund, J.; Grant, B.R.; Grant, P.R.; Webster, M.T.; Andersson, L. Adaptive radiation of Darwin’s finches revisited using whole genome sequencing. BioEssays 2016, 38, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Dasmahapatra, K.K.; Walters, J.R.; Briscoe, A.D.; Davey, J.W.; Whibley, A.; Nadeau, N.J.; Zimin, A.V.; Hughes, D.S.T.; Ferguson, L.C.; Martin, S.H.; et al. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 2012, 487, 94–98. [Google Scholar] [CrossRef] [Green Version]
- Seehausen, O. Hybridization and adaptive radiation. Trends Ecol. Evol. 2004, 19, 198–207. [Google Scholar] [CrossRef]
- Abzhanov, A.; Kuo, W.P.; Hartmann, C.; Grant, B.R.; Grant, P.R.; Tabin, C.J. The calmodulin pathway and evolution of elongated beak morphology in Darwin’s finches. Nature 2006, 442, 563–567. [Google Scholar] [CrossRef]
- Abzhanov, A.; Protas, M.; Grant, B.R.; Grant, P.R.; Tabin, C.J. Bmp4 and morphological variation of beaks in Darwin’s finches. Science 2004, 305, 1462–1465. [Google Scholar] [CrossRef] [Green Version]
- Lawson, L.P.; Petren, K. The adaptive genomic landscape of beak morphology in Darwin’s finches. Mol. Ecol. 2017, 26, 4978–4989. [Google Scholar] [CrossRef]
- Kruimel, J.H. Verzeichnis der von Herrn, E.C. Abendanon in Celebes gesammelten Süsswasser-Mollusken. Bijdr. Tot Dierkd. 1913, 19, 217–235. [Google Scholar]
- von Rintelen, T.; Wilson, A.B.; Meyer, A.; Glaubrecht, M. Escalation and trophic specialization drive adaptive radiation of freshwater gastropods in ancient lakes on Sulawesi, Indonesia. Proc. R. Soc. London B 2004, 271, 2541–2549. [Google Scholar] [CrossRef] [PubMed]
- von Rintelen, T.; von Rintelen, K.; Glaubrecht, M. The Species Flocks of the Viviparous Freshwater Gastropod Tylomelania (Mollusca: Cerithioidea: Pachychilidae) in the Ancient Lakes of Sulawesi, Indonesia: The Role of Geography, Trophic Morphology and Color as Driving Forces in Adaptive Radiation. In Evolution in Action; Glaubrecht, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 485–512. [Google Scholar]
- Von Rintelen, T.; Glaubrecht, M. Anatomy of an adaptive radiation: A unique reproductive strategy in the endemic freshwater gastropod Tylomelania (Cerithioidea: Pachychilidae) on Sulawesi, Indonesia and its biogeographical implications. Biol. J. Linn. Soc. 2005, 85, 513–542. [Google Scholar] [CrossRef] [Green Version]
- Glaubrecht, M.; von Rintelen, T. The species flocks of lacustrine gastropods: Tylomelania on Sulawesi as models in speciation and adaptive radiation. Hydrobiologia 2008, 615, 181–199. [Google Scholar] [CrossRef]
- Hilgers, L.; Grau, J.H.; Pfaender, J.; von Rintelen, T. The complete mitochondrial genome of the viviparous freshwater snail Tylomelania sarasinorum (Caenogastropoda: Cerithioidea). Mitochondrial DNA Part B 2016, 1, 389–390. [Google Scholar] [CrossRef] [Green Version]
- Mackenstedt, U.; Märkel, K. Experimental and comparative morphology of radula renewal in pulmonates (Mollusca, Gastropoda). Zoomorphology 1987, 107, 209–239. [Google Scholar] [CrossRef]
- Padilla, D.K. Inducible phenotypic plasticity of the radula in Lacuna (Gastropoda: Littorinidae). Veliger 1998, 4, 201–204. [Google Scholar]
- von Rintelen, T.; Bouchet, P.; Glaubrecht, M. Ancient lakes as hotspots of diversity: A morphological review of an endemic species flock of Tylomelania (Gastropoda: Cerithioidea: Pachychilidae) in the Malili lake system on Sulawesi, Indonesia. Hydrobiologia 2007, 592, 11–94. [Google Scholar] [CrossRef]
- Rohlf, F.J. TpsDig2; Department of Ecology and Evolution, State Universityof New York, Stony Brook: New York, NY, USA, 2006. [Google Scholar]
- Adams, D.C.; Otárola-Castillo, E. Geomorph: An R package for the collection and analysis of geometric morphometric shape data. Methods Ecol. Evol. 2013, 4, 393–399. [Google Scholar] [CrossRef]
- Holznagel, W.E. A nondestructive method for cleaning gastropod radulae from frozen, alcohol-fixed, or dried material. Am. Malacol. Bull. 1998, 14, 181–183. [Google Scholar]
- Rasband, W.U. ImageJ. U.S. Available online: https://imagej.nih.gov/ij/ (accessed on 14 May 2022).
- Hilgers, L.; Hartmann, S.; Hofreiter, M.; von Rintelen, T. Novel genes, ancient genes, and gene co-option contributed to the genetic basis of the radula, a molluscan innovation. Mol. Biol. Evol. 2018, 35, 1638–1652. [Google Scholar] [CrossRef] [PubMed]
- Sultan, M.; Dökel, S.; Amstislavskiy, V.; Wuttig, D.; Sültmann, H.; Lehrach, H.; Yaspo, M.L. A simple strand-specific RNA-Seq library preparation protocol combining the Illumina TruSeq RNA and the dUTP methods. Biochem. Biophys. Res. Commun. 2012, 422, 643–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, N.A.; Fass, J.N. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files 2011. Available online: https://github.com/najoshi/sickle (accessed on 14 May 2022).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [Green Version]
- Kofler, R.; Pandey, R.V.; Schlötterer, C. PoPoolation2: Identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics 2011, 27, 3435–3436. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Roesti, M.; Salzburger, W.; Berner, D. Uninformative polymorphisms bias genome scans for signatures of selection. BMC Evol. Biol. 2012, 12, 94. [Google Scholar] [CrossRef] [Green Version]
- Konczal, M.; Koteja, P.; Stuglik, M.T.; Radwan, J.; Babik, W. Accuracy of allele frequency estimation using pooled RNA-Seq. Mol. Ecol. Resour. 2014, 14, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Konczal, M.; Koteja, P.; Orlowska-Feuer, P.; Radwan, J.; Sadowska, E.T.; Babik, W. Genomic response to selection for predatory behavior in a mammalian model of adaptive radiation. Mol. Biol. Evol. 2016, 33, 2429–2440. [Google Scholar] [CrossRef] [PubMed]
- Kozak, G.M.; Brennan, R.S.; Berdan, E.L.; Fuller, R.C.; Whitehead, A. Functional and population genomic divergence within and between two species of killifish adapted to different osmotic niches. Evolution 2014, 68, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Schlötterer, C.; Tobler, R.; Kofler, R.; Nolte, V. Sequencing pools of individuals—Mining genome-wide polymorphism data without big funding. Nat. Rev. Genet. 2014, 15, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Westram, A.M.; Galindo, J.; Rosenblad, M.A.; Grahame, J.W.; Panova, M.; Butlin, R.K. Do the same genes underlie parallel phenotypic divergence in different Littorina saxatilis populations? Mol. Ecol. 2014, 23, 4603–4616. [Google Scholar] [CrossRef] [Green Version]
- Harney, E.; Dubief, B.; Boudry, P.; Basuyaux, O.; Schilhabel, M.B.; Huchette, S.; Paillard, C.; Nunes, F.L.D. De novo assembly and annotation of the European abalone Haliotis tuberculata transcriptome. Mar. Genom. 2016, 28, 11–16. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, A.L.; Wollesen, T.; Kristof, A.; Scherholz, M.; Redl, E.; Todt, C.; Bleidorn, C.; Wanninger, A. Comparative transcriptomics enlarges the toolkit of known developmental genes in mollusks. BMC Genom. 2016, 17, 905. [Google Scholar] [CrossRef] [Green Version]
- Mack, K.L.; Nachman, M.W. Gene Regulation and Speciation. Trends Genet. 2017, 33, 68–80. [Google Scholar] [CrossRef] [Green Version]
- Rougeux, C.; Gagnaire, P.; Praebel, K.; Seehausen, O.; Bernatchez, L. Polygenic selection drives the evolution of convergent transcriptomic landscapes across continents within a Nearctic sister-species complex. Mol. Ecol. 2019, 28, 4388–4403. [Google Scholar] [CrossRef]
- Uebbing, S.; Künstner, A.; Mäkinen, H.; Backström, N.; Bolivar, P.; Burri, R.; Dutoit, L.; Mugal, C.F.; Nater, A.; Aken, B.; et al. Divergence in gene expression within and between two closely related flycatcher species. Mol. Ecol. 2016, 25, 2015–2028. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, M.; Schrey, A.W.; Richards, C.L. Ten years of transcriptomics in wild populations: What have we learned about their ecology and evolution? Mol. Ecol. 2015, 24, 710–725. [Google Scholar] [CrossRef]
- El Taher, A.; Böhne, A.; Boileau, N.; Ronco, F.; Indermaur, A.; Widmer, L.; Salzburger, W. Gene expression dynamics during rapid organismal diversification in African cichlid fishes. Nat. Ecol. Evol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Moon, S. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003, 13, 13–22. [Google Scholar] [CrossRef]
- Redmond, L.; Ghosh, A. The role of Notch and Rho GTPase signaling in the control of dendritic development. Curr. Opin. Neurobiol. 2001, 11, 111–117. [Google Scholar] [CrossRef]
- Polacheck, W.J.; Kutys, M.L.; Yang, J.; Eyckmans, J.; Wu, Y.; Vasavada, H.; Hirschi, K.K.; Chen, C.S. A non-canonical Notch complex regulates adherens junctions and vascular barrier function. Nature 2017, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Richards, G.S.; Degnan, B.M. The dawn of developmental signaling in the metazoa. Cold Spring Harb. Symp. Quant. Biol. 2009, 74, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Heller, E.; Fuchs, E. Tissue patterning and cellular mechanics. J. Cell Biol. 2015, 211, 219–231. [Google Scholar] [CrossRef]
- Roberts, R.B.; Hu, Y.; Albertson, R.C.; Kocher, T.D. Craniofacial divergence and ongoing adaptation via the hedgehog pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 13194–13199. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Albertson, R.C. Baby fish working out: An epigenetic source of adaptive variation in the cichlid jaw. Proc. R. Soc. B Biol. Sci. 2017, 284, 20171018. [Google Scholar] [CrossRef]
- Hu, Y.; Albertson, R.C. Hedgehog signaling mediates adaptive variation in a dynamic functional system in the cichlid feeding apparatus. Proc. Natl. Acad. Sci. USA 2014, 111, 8530–8534. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Ríos, P.; Guerrero, I.; González-Reyes, A. Cytoneme-mediated delivery of hedgehog regulates the expression of bone morphogenetic proteins to maintain germline stem cells in Drosophila. PLoS Biol. 2012, 10, e1001298. [Google Scholar] [CrossRef] [Green Version]
- Cicconardi, F.; Marcatili, P.; Arthofer, W.; Schlick-Steiner, B.C.; Steiner, F.M. Positive diversifying selection is a pervasive adaptive force throughout the Drosophila radiation. Mol. Phylogenet. Evol. 2017, 112, 230–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albertson, R.C.; Kocher, T.D. Genetic and developmental basis of cichlid trophic diversity. Heredity 2006, 97, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albertson, R.C.; Streelman, J.T.; Kocher, T.D. Genetic basis of adaptive shape differences in the cichlid head. J. Hered. 2003, 94, 291–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feder, J.L.; Egan, S.P.; Nosil, P. The genomics of speciation-with-gene-flow. Trends Genet. 2012, 28, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Seehausen, O.; Butlin, R.K.; Keller, I.; Wagner, C.E.; Boughman, J.W.; Hohenlohe, P.A.; Peichel, C.L.; Saetre, G.-P.; Bank, C.; Brännström, A.; et al. Genomics and the origin of species. Nat. Rev. Genet. 2014, 15, 176–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Rintelen, T.; Glaubrecht, M. New discoveries in old lakes: Three new species of Tylomelania Sarasin & Sarasin, 1897 (Gastropoda: Cerithioidea: Pachychilidae) from the Malili lake system on Sulawesi, Indonesia. J. Molluscan Stud. 2003, 69, 3–17. [Google Scholar]
- Stern, D.L.; Orgogozo, V. The loci of evolution: How predictable is genetic evolution? Evolution 2008, 62, 2155–2177. [Google Scholar] [CrossRef] [Green Version]
- Stern, D.L.; Orgogozo, V. Is genetic evolution predictable? Science 2009, 323, 746–751. [Google Scholar] [CrossRef] [Green Version]
- Jones, F.C.; Grabherr, M.G.; Chan, Y.F.; Russell, P.; Mauceli, E.; Johnson, J.; Swofford, R.; Pirun, M.; Zody, M.C.; White, S.; et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 2012, 484, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Khaitovich, P.; Weiss, G.; Lachmann, M.; Hellmann, I.; Enard, W.; Muetzel, B.; Wirkner, U.; Ansorge, W.; Pääbo, S. A neutral model of transcriptome evolution. PLoS Biol. 2004, 2, e132. [Google Scholar] [CrossRef]
- Catalán, A.; Briscoe, A.D.; Höhna, S. Drift and directional selection are the evolutionary forces driving gene expression divergence in eye and brain tissue of Heliconius butterflies. Genetics 2019, 213, 581–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakrzewski, A.C.; Weigert, A.; Helm, C.; Adamski, M.; Adamska, M.; Bleidorn, C.; Raible, F.; Hausen, H. Early divergence, broad distribution, and high diversity of animal chitin synthases. Genome Biol. Evol. 2014, 6, 316–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, J.I.; Marques, D.A.; Mwaiko, S.; Wagner, C.E.; Excoffier, L.; Seehausen, O. Ancient hybridization fuels rapid cichlid fish adaptive radiations. Nat. Commun. 2017, 8, 14363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enciso-Romero, J.; Pardo-Díaz, C.; Martin, S.H.; Arias, C.F.; Linares, M.; McMillan, W.O.; Jiggins, C.D.; Salazar, C. Evolution of novel mimicry rings facilitated by adaptive introgression in tropical butterflies. Mol. Ecol. 2017, 26, 5160–5172. [Google Scholar] [CrossRef]
- Lamichhaney, S.; Han, F.; Webster, M.T.; Andersson, L.; Grant, B.R.; Grant, P.R. Rapid hybrid speciation in Darwin’s finches. Science 2017, 4593, eaao4593. [Google Scholar] [CrossRef] [Green Version]
- Marques, D.A.; Meier, J.I.; Seehausen, O. A Combinatorial View on Speciation and Adaptive Radiation. Trends Ecol. Evol. 2019, 34, 531–544. [Google Scholar] [CrossRef] [Green Version]
- Fish, J.L. Evolvability of the vertebrate craniofacial skeleton. Semin. Cell Dev. Biol. 2017, 91, 13–22. [Google Scholar] [CrossRef]
- Pires-daSilva, A.; Sommer, R.J. The evolution of signalling pathways in animal development. Nat. Rev. Genet. 2003, 4, 39–49. [Google Scholar] [CrossRef]
- Jiggins, C.D.; Wallbank, R.W.R.; Hanly, J.J. Waiting in the wings: What can we learn about gene co-option from the diversification of butterfly wing patterns? Philos. Trans. R. Soc. B 2016, 372, 20150485. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hilgers, L.; Hartmann, S.; Pfaender, J.; Lentge-Maaß, N.; Marwoto, R.M.; von Rintelen, T.; Hofreiter, M. Evolutionary Divergence and Radula Diversification in Two Ecomorphs from an Adaptive Radiation of Freshwater Snails. Genes 2022, 13, 1029. https://doi.org/10.3390/genes13061029
Hilgers L, Hartmann S, Pfaender J, Lentge-Maaß N, Marwoto RM, von Rintelen T, Hofreiter M. Evolutionary Divergence and Radula Diversification in Two Ecomorphs from an Adaptive Radiation of Freshwater Snails. Genes. 2022; 13(6):1029. https://doi.org/10.3390/genes13061029
Chicago/Turabian StyleHilgers, Leon, Stefanie Hartmann, Jobst Pfaender, Nora Lentge-Maaß, Ristiyanti M. Marwoto, Thomas von Rintelen, and Michael Hofreiter. 2022. "Evolutionary Divergence and Radula Diversification in Two Ecomorphs from an Adaptive Radiation of Freshwater Snails" Genes 13, no. 6: 1029. https://doi.org/10.3390/genes13061029
APA StyleHilgers, L., Hartmann, S., Pfaender, J., Lentge-Maaß, N., Marwoto, R. M., von Rintelen, T., & Hofreiter, M. (2022). Evolutionary Divergence and Radula Diversification in Two Ecomorphs from an Adaptive Radiation of Freshwater Snails. Genes, 13(6), 1029. https://doi.org/10.3390/genes13061029