
Major Histocompatibility Complex (MHC) Diversity of the Reintroduction Populations of Endangered Przewalski’s Horse

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and PCR
2.3. SSCP Analysis and Cloning
2.4. Statistical Analysis
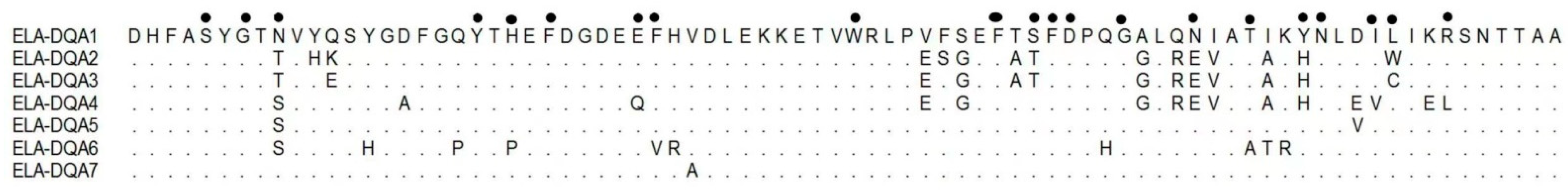
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bernatchez, L.; Landry, C. MHC studies in nonmodel vertebrates: What have we learned about natural selection in 15 years? J. Evol. Biol. 2003, 16, 363–377. [Google Scholar] [CrossRef]
- Rhodes, D.A.; Trowsdale, J. Genetics and molecular genetics of the MHC. Rev. Immunogenet. 1999, 1, 21–31. [Google Scholar]
- Radwan, J.; Babik, W.; Kaufman, J.; Lenz, T.L.; Winternitz, J. Advances in the Evolutionary Understanding of MHC Polymorphism. Trends Genet. 2020, 36, 298–311. [Google Scholar] [CrossRef]
- Martinsohn, J.T.; Sousa, A.B.; Guethlein, L.A.; Howard, J.C. The gene conversion hypothesis of MHC evolution: A review. Immunogenetics 1999, 50, 168–200. [Google Scholar] [CrossRef]
- Eizaguirre, C.; Lenz, T.L.; Kalbe, M.; Milinski, M. Rapid and adaptive evolution of MHC genes under parasite selection in experimental vertebrate populations. Nat. Commun. 2012, 3, 621. [Google Scholar] [CrossRef]
- Sutton, J.T.; Nakagawa, S.; Robertson, B.C.; Jamieson, I.G. Data from: Disentangling the roles of natural selection and genetic drift in shaping variation at MHC immunity genes. Mol. Ecol. 2011, 20, 4408–4420. [Google Scholar] [CrossRef]
- Kamiya, T.; O’Dwyer, K.; Westerdahl, H.; Senior, A.; Nakagawa, S. Data from: A quantitative review of MHC-based mating preference: The role of diversity and dissimilarity. Mol. Ecol. 2014, 23, 5151–5163. [Google Scholar] [CrossRef]
- Winternitz, J.J.A.; Abbate, J.L.; Huchard, E.; Havlicek, J.; Garamszegi, L.Z. Patterns of MHC-dependent mate selection in humans and nonhuman primates: A meta-analysis. Mol. Ecol. 2016, 26, 668–688. [Google Scholar] [CrossRef]
- Ujvari, B.; Belov, K. Major Histocompatibility Complex (MHC) Markers in Conservation Biology. Int. J. Mol. Sci. 2011, 12, 5168–5186. [Google Scholar] [CrossRef]
- Burger, D.; Thomas, S.; Aepli, H.; Dreyer, M.; Fabre, G.; Marti, E.; Sieme, H.; Robinson, M.R.; Wedekind, C. Major histocompatibility complex-linked social signalling affects female fertility. Proc. R. Soc. B Biol. Sci. 2017, 284, 20171824. [Google Scholar] [CrossRef]
- Gupta, P.; Robin, V.V.; Dharmarajan, G. Towards a more healthy conservation paradigm: Integrating disease and molecular ecology to aid biological conservation. J. Genet. 2020, 99, 65. [Google Scholar] [CrossRef]
- Morris, K.; Austin, J.; Belov, K. Low major histocompatibility complex diversity in the Tasmanian devil predates European settlement and may explain susceptibility to disease epidemics. Biol. Lett. 2013, 9, 20120900. [Google Scholar] [CrossRef]
- Morris, K.M.; Wright, B.; Grueber, C.E.; Hogg, C.; Belov, K. Data from: Lack of genetic diversity across diverse immune genes in an endangered mammal, the Tasmanian devil (Sarcophilus harrisii). Mol. Ecol. 2015, 24, 3860–3872. [Google Scholar] [CrossRef]
- Belov, K. The role of the Major Histocompatibility Complex in the spread of contagious cancers. Mamm. Genome 2010, 22, 83–90. [Google Scholar] [CrossRef]
- Hohenlohe, P.A.; McCallum, H.I.; Jones, M.E.; Lawrance, M.F.; Hamede, R.K.; Storfer, A. Conserving adaptive potential: Lessons from Tasmanian devils and their transmissible cancer. Conserv. Genet. 2019, 20, 81–87. [Google Scholar] [CrossRef]
- Piertney, S.B.; Oliver, M.K. The evolutionary ecology of the major histocompatibility complex. Heredity 2005, 96, 7–21. [Google Scholar] [CrossRef]
- Lau, A.N.; Peng, L.; Goto, H.; Chemnick, L.; Ryder, O.A.; Makova, K.D. Horse Domestication and Conservation Genetics of Przewalski’s Horse Inferred from Sex Chromosomal and Autosomal Sequences. Mol. Biol. Evol. 2008, 26, 199–208. [Google Scholar] [CrossRef]
- Liu, G.; Xu, C.-Q.; Cao, Q.; Zimmermann, W.; Songer, M.; Zhao, S.-S.; Li, K.; Hu, D.-F. Mitochondrial and pedigree analysis in Przewalski’s horse populations: Implications for genetic management and reintroductions. Mitochondrial DNA 2013, 25, 313–318. [Google Scholar] [CrossRef]
- Oakenfull, E.A.; Ryder, O.A. Mitochondrial control region and 12S rRNA variation in Przewalski’s horse (Equus przewalskii). Anim. Genet. 1998, 29, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Shafer, A.B.; Zimmermann, W.; Hu, D.; Wang, W.; Chu, H.; Cao, J.; Zhao, C. Evaluating the reintroduction project of Przewalski’s horse in China using genetic and pedigree data. Biol. Conserv. 2014, 171, 288–298. [Google Scholar] [CrossRef]
- Breen, M.; Downs, P.; Irvin, Z.; Bell, K. Intrageneric amplification of horse microsatellite markers with emphasis on the Przewalski’s horse (E. pmewalskii). Anim. Genet. 2009, 25, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Der Sarkissian, C.; Ermini, L.; Schubert, M.; Yang, M.A.; Librado, P.; Fumagalli, M.; Jónsson, H.; Bar-Gal, G.K.; Albrechtsen, A.; Vieira, F.G.; et al. Evolutionary Genomics and Conservation of the Endangered Przewalski’s Horse. Curr. Biol. 2015, 25, 2577–2583. [Google Scholar] [CrossRef] [PubMed]
- Gaunitz, C.; Fages, A.; Hanghøj, K.; Albrechtsen, A.; Khan, N.; Schubert, M.; Seguin-Orlando, A.; Owens, I.J.; Felkel, S.; Bignon-Lau, O.; et al. Ancient genomes revisit the ancestry of domestic and Przewalski’s horses. Science 2018, 360, 111–114. [Google Scholar] [CrossRef]
- Hedrick, P.W.; Parker, K.M.; Miller, E.L.; Miller, P.S. Major histocompatibility complex variation in the endangered Przewalski’s horse. Genetics 1999, 152, 1701–1710. [Google Scholar] [CrossRef]
- Bowling, A.T.; Zimmermann, W.; Ryder, O.; Penado, C.; Peto, S.; Chemnick, L.; Yasinetskaya, N.; Zharkikh, T. Genetic variation in Przewalski’s horses, with special focus on the last wild caught mare, 231 Orlitza III. Cytogenet. Genome Res. 2003, 102, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Isolation of High-molecular-weight DNA from Mammalian Cells Using Proteinase K and Phenol. CSH Protoc. 2006. [Google Scholar] [CrossRef] [PubMed]
- Natkaniec, M.; Potaczek, D.P.; Sanak, M. Single-stranded conformation polymorphism (SSCP)-driven indirect sequencing in detection of short deletion. Mol. Biol. Rep. 2008, 36, 1545–1547. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Rousset, F. genepop’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef]
- Paliakasis, K.; Routsias, J.; Petratos, K.; Ouzounis, C.; Kokkinidis, M.; Papadopoulos, G.K. Novel Structural Features of the Human Histocompatibility Molecules HLA-DQ as Revealed by Modeling Based on the Published Structure of the Related Molecule HLA-DR. J. Struct. Biol. 1996, 117, 145–163. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.-H.; Zhu, L.; Wu, H.; Fang, S.-G. Major histocompatibility complex class II variation in the giant panda (Ailuropoda melanoleuca). Mol. Ecol. 2006, 15, 2441–2450. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Huang, K.; Zhang, B.; Dunn, D.W.; Chen, D.; Li, F.; Qi, X.; Guo, S.; Li, B. High polymorphism in MHC-DRB genes in golden snub-nosed monkeys reveals balancing selection in small, isolated populations. BMC Evol. Biol. 2018, 18, 29. [Google Scholar] [CrossRef] [PubMed]
- Kaczensky, P.; Ganbaatar, O.; Von Wehrden, H.; Enksaikhan, N.; Lkhagvasuren, D.; Walzer, C. Przewalski’s Horse (Equus ferus przewalskii) Re-introduction in the Great Gobi B Strictly Protected Area: From Species to Ecosystem Conservation. Mong. J. Biol. Sci. 2007, 5, 13–18. [Google Scholar] [PubMed]
- Osada, N. Genetic diversity in humans and non-human primates and its evolutionary consequences. Genes Genet. Syst. 2015, 90, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zheng, Y.; Zhao, J.; Yao, M. Low genetic diversity in a critically endangered primate: Shallow evolutionary history or recent population bottleneck? BMC Evol. Biol. 2019, 19, 134. [Google Scholar] [CrossRef]
- Lehnen, L.; Jan, P.; Besnard, A.; Fourcy, D.; Kerth, G.; Biedermann, M.; Nyssen, P.; Schorcht, W.; Petit, E.J.; Puechmaille, S.J. Genetic diversity in a long-lived mammal is explained by the past’s demographic shadow and current connectivity. Mol. Ecol. 2021, 30, 5048–5063. [Google Scholar] [CrossRef]
- Jaworska, J.; Ropka-Molik, K.; Wocławek-Potocka, I.; Siemieniuch, M.J. Inter- and intrabreed diversity of the major histocompatibility complex (MHC) in primitive and draft horse breeds. PLoS ONE 2020, 15, e0228658. [Google Scholar] [CrossRef]
- Viļuma, A.; Mikko, S.; Hahn, D.; Skow, L.; Andersson, G.; Bergström, T.F. Genomic structure of the horse major histocompatibility complex class II region resolved using PacBio long-read sequencing technology. Sci. Rep. 2017, 7, 45518. [Google Scholar] [CrossRef]
- Tallmadge, R.L.; Campbell, J.A.; Miller, D.C.; Antczak, D.F. Analysis of MHC class I genes across horse MHC haplotypes. Immunogenetics 2010, 62, 159–172. [Google Scholar] [CrossRef]
- Rüegg, S.R.; Torgerson, P.R.; Doherr, M.G.; Deplazes, P.; Böse, R.; Robert, N.; Walzer, C. Equine piroplasmoses at the reintroduction site of the przewalski’s horse (Equus ferus przewalskii) in mongolia. J. Wildl. Dis. 2006, 42, 518–526. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baptista, C.J.; Sós, E.; Szabados, T.; Kerekes, V.; de Carvalho, L.M. Intestinal parasites in Przewalski’s horses (Equus ferus przewalskii): A field survey at the Hortobágy National Park, Hungary. J. Helminthol. 2021, 95, e39. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Chao, Y.; Zhang, B.; Wang, C.; Qi, Y.; Ente, M.; Zhang, D.; Li, K.; Mok, K.M. Effects of Gasterophilus pecorum infestation on the intestinal microbiota of the rewilded Przewalski’s horses in China. PLoS ONE 2021, 16, e0251512. [Google Scholar] [CrossRef] [PubMed]
- Kleinman-Ruiz, D.; Soriano, L.; Casas-Marce, M.; Szychta, C.; Sánchez, I.; Fernández, J.; Godoy, J.A. Genetic evaluation of the Iberian lynx ex situ conservation programme. Heredity 2019, 123, 647–661. [Google Scholar] [CrossRef]
- Zhang, S.; Li, C.; Li, Y.; Chen, Q.; Hu, D.; Cheng, Z.; Wang, X.; Shan, Y.; Bai, J.; Liu, G. Genetic Differentiation of Reintroduced Père David’s Deer (Elaphurus davidianus) Based on Population Genomics Analysis. Front. Genet. 2021, 12, 705337. [Google Scholar] [CrossRef]
- Flesch, E.P.; Graves, T.A.; Thomson, J.M.; Proffitt, K.M.; White, P.J.; Stephenson, T.R.; Garrott, R.A. Evaluating wildlife translocations using genomics: A bighorn sheep case study. Ecol. Evol. 2020, 10, 13687–13704. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Population | Sample Type | Sample Size | Sampling Year |
|---|---|---|---|
| PHB | Hair | 2 | 2009 |
| Blood | 9 | 2009 | |
| EAB | Faces | 20 | 2015 |
| Population | ELA- DQA1 | ELA- DQA2 | ELA- DQA3 | ELA- DQA4 | ELA- DQA5 | ELA- DQA6 | ELA- DQA7 | p Value |
|---|---|---|---|---|---|---|---|---|
| PHB | 0.00 | 0.23 | 0.36 | 0.36 | 0.00 | 0.05 | 0.00 | 0.0033 |
| EAB | 0.65 | 0.00 | 0.20 | 0.00 | 0.13 | 0.00 | 0.03 | 0.00 |
| Population | Ho | He | PIC | Ne |
|---|---|---|---|---|
| PHB | 0.32 | 0.69 | 0.62 | 3.14 |
| EAB | 0.47 | 0.54 | 0.42 | 1.86 |
| Average | 0.39 | 0.61 | 0.52 | 2.5 |
| Kimura 2-Parameter | Amino Acid Genetic Distance (Poisson Correction) | ||||
|---|---|---|---|---|---|
| All Sites | ABS | Non-ABS | All Sites | ABS | Non-ABS |
| 8.5 | 19.7 | 5.5 | 17.2 | 38.7 | 11.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, Y.; Liu, G.; Zhao, S.; Li, K.; Zhang, D.; Liu, S.; Hu, D. Major Histocompatibility Complex (MHC) Diversity of the Reintroduction Populations of Endangered Przewalski’s Horse. Genes 2022, 13, 928. https://doi.org/10.3390/genes13050928
Tang Y, Liu G, Zhao S, Li K, Zhang D, Liu S, Hu D. Major Histocompatibility Complex (MHC) Diversity of the Reintroduction Populations of Endangered Przewalski’s Horse. Genes. 2022; 13(5):928. https://doi.org/10.3390/genes13050928
Chicago/Turabian StyleTang, Yongqing, Gang Liu, Shasha Zhao, Kai Li, Dong Zhang, Shuqiang Liu, and Defu Hu. 2022. "Major Histocompatibility Complex (MHC) Diversity of the Reintroduction Populations of Endangered Przewalski’s Horse" Genes 13, no. 5: 928. https://doi.org/10.3390/genes13050928
APA StyleTang, Y., Liu, G., Zhao, S., Li, K., Zhang, D., Liu, S., & Hu, D. (2022). Major Histocompatibility Complex (MHC) Diversity of the Reintroduction Populations of Endangered Przewalski’s Horse. Genes, 13(5), 928. https://doi.org/10.3390/genes13050928