
Prominent Follicular Keratosis in Multiple Intestinal Atresia with Combined Immune Deficiency Caused by a TTC7A Homozygous Mutation

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
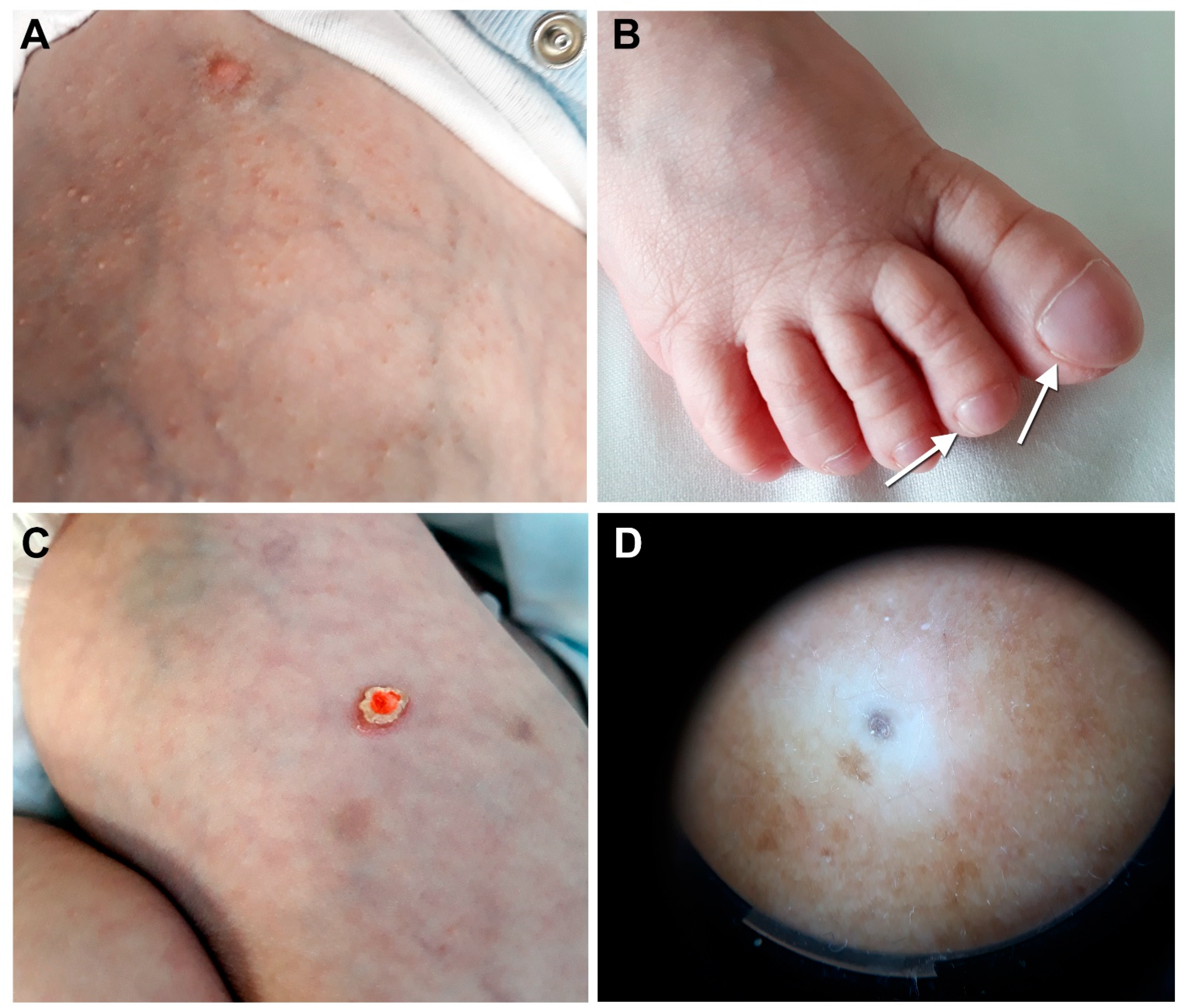
3.1. Case Report
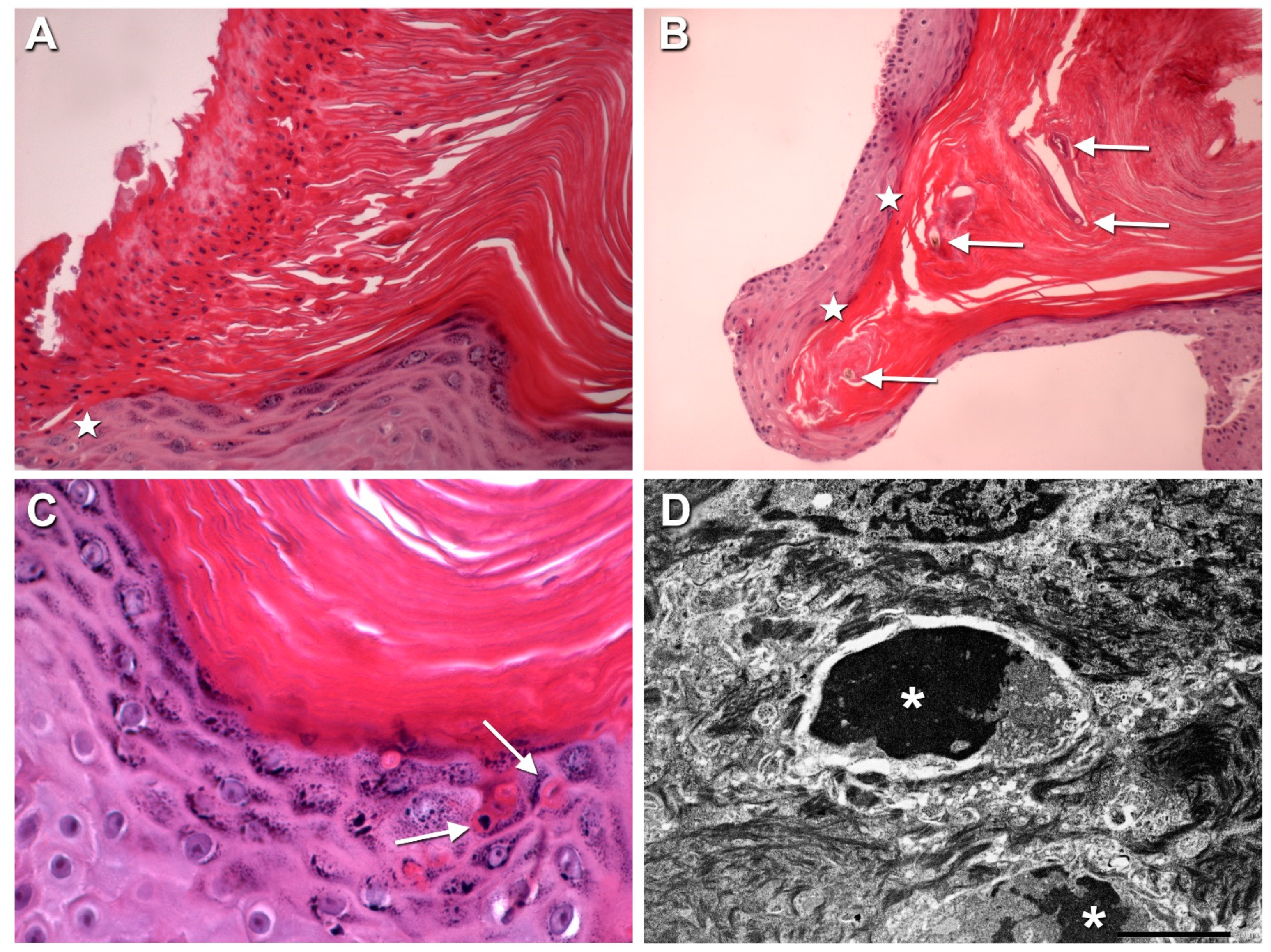
3.2. Histopathological and Ultrastructural Findings
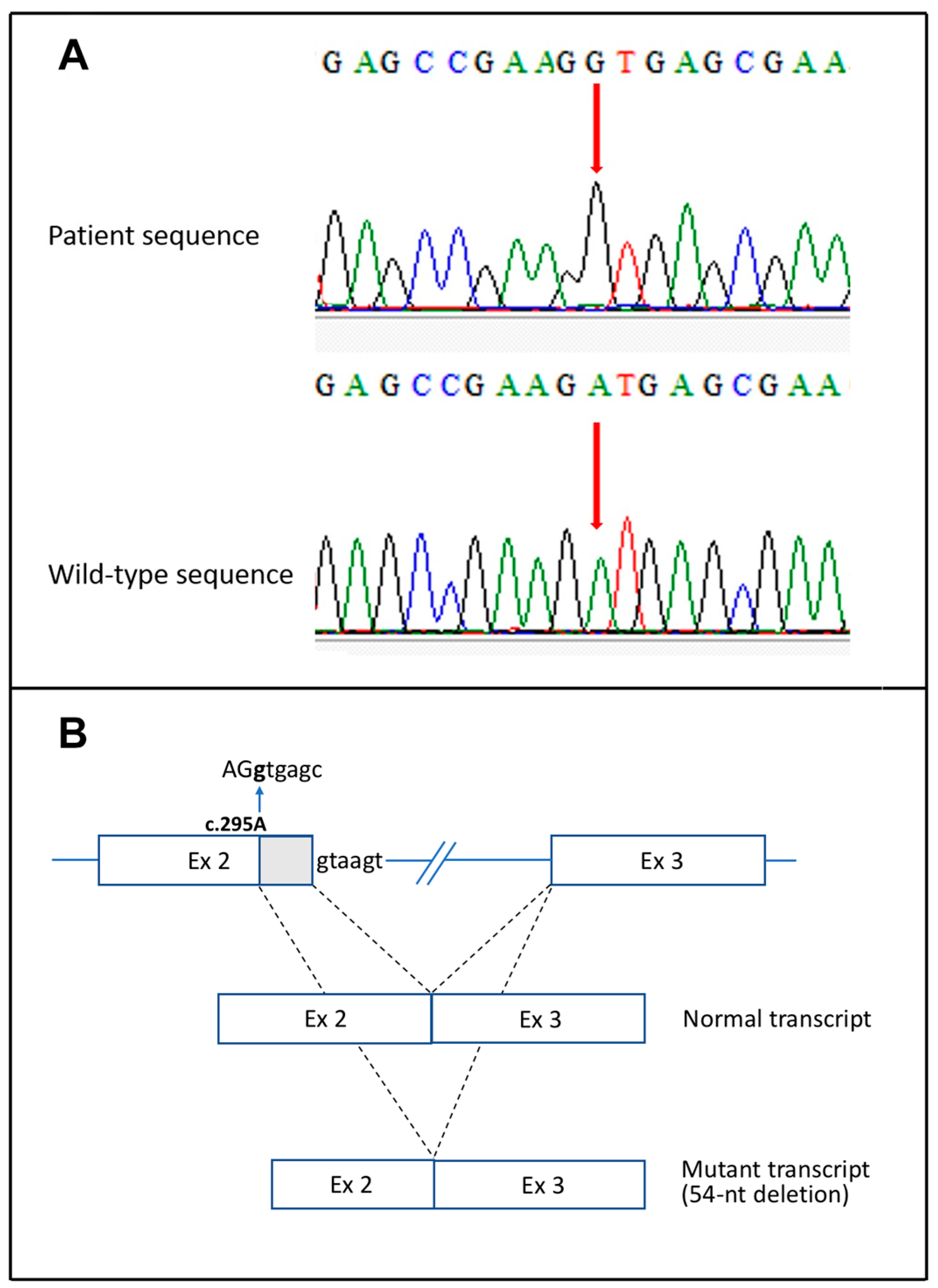
3.3. Mutation Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Samuels, M.E.; Majewski, J.; Alirezaie, N.; Fernandez, I.; Casals, F.; Patey, N.; Decaluwe, H.; Gosselin, I.; Haddad, E.; Hodgkinson, A.; et al. Exome sequencing identifies mutations in the gene TTC7A in French-Canadian cases with hereditary multiple intestinal atresia. J. Med. Genet. 2013, 50, 324–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Giliani, S.; Lanzi, G.; Mias, G.I.; Lonardi, S.; Dobbs, K.; Manis, J.; Im, H.; Gallagher, J.E.; Phanstiel, D.H.; et al. Whole-exome sequencing identifies tetratricopeptide repeat domain 7A (TTC7A) mutations for combined immunodeficiency with intestinal atresias. J. Allergy Clin. Immunol. 2013, 132, 656–664.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jardine, S.; Dhingani, N.; Muise, A.M. TTC7A: Steward of intestinal health. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 555–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Daher, M.T.; Lemale, J.; Bruneau, J.; Leveau, C.; Guerin, F.; Lambert, N.; Diana, J.S.; Neven, B.; Sepulveda, F.E.; Coulomb-L’Hermine, A.; et al. Chronic intestinal pseudo-obstruction and lymphoproliferative syndrome as a novel phenotype associated with tetratricopeptide repeat domain 7A deficiency. Front. Immunol. 2019, 10, 2592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemoine, R.; Pachlopnik-Schmid, J.; Farin, H.F.; Bigorgne, A.; Debré, M.; Sepulveda, F.; Héritier, S.; Lemale, J.; Talbotec, C.; Rieux-Laucat, F.; et al. Immune deficiency-related enteropathy-lymphocytopenia-alopecia syndrome results from tetratricopeptide repeat domain 7A deficiency. J. Allergy Clin. Immunol. 2014, 134, 1354–1364.e6. [Google Scholar] [CrossRef] [PubMed]
- Bigorgne, A.E.; Farin, H.F.; Lemoine, R.; Mahlaoui, N.; Lambert, N.; Gil, M.; Schulz, A.; Philippet, P.; Schlesser, P.; Abrahamsen, T.G.; et al. TTC7A mutations disrupt intestinal epithelial picobasal polarity. J. Clin. Investig. 2014, 124, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Leclerc-Mercier, S.; Lemoine, R.; Bigorgne, A.E.; Sepulveda, F.; Leveau, C.; Fischer, A.; Mahlaoui, N.; Hadj-Rabia, S.; de Saint Basile, G. Ichthyosis as the dermatological phenotype associated with TTC7A mutations. Br. J. Dermatol. 2016, 175, 1061–1064. [Google Scholar] [CrossRef] [PubMed]
- Alipour Tehrany, Y.; Marois, L.; Colmant, C.; Marchand, V.; Kokta, V.; Coulombe, J.; Marcoux, D.; Haddad, E.; McCuaig, C. Refractory pruritus responds to dupilumab in a patient with TTC7A mutation. JAAD Case Rep. 2020, 8, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Lien, R.; Lin, Y.F.; Lai, M.W.; Weng, H.Y.; Wu, R.C.; Jaing, T.H.; Huang, J.L.; Tsai, S.F.; Lee, W.I. Novel mutations of the Tetratricopeptide Repeat Domain 7A gene and phenotype/genotype comparison. Front. Immunol. 2017, 8, 1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avitzur, Y.; Guo, C.; Mastropaolo, L.A.; Bahrami, E.; Chen, H.; Zhao, Z.; Elkadri, A.; Dhillon, S.; Murchie, R.; Fattouh, R.; et al. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology 2014, 146, 1028–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mou, W.; Yang, S.; Guo, R.; Fu, L.; Zhang, L.; Guo, W.; Du, J.; He, J.; Ren, Q.; Hao, C.; et al. A novel homozygous TTC7A missense mutation results in familial multiple intestinal atresia and combined immunodeficiency. Front. Immunol. 2021, 12, 759308. [Google Scholar] [CrossRef] [PubMed]
- Kammermeier, J.; Lucchini, G.; Pai, S.Y.; Worth, A.; Rampling, D.; Amrolia, P.; Silva, J.; Chiesa, R.; Rao, K.; Noble-Jamieson, G.; et al. Stem cell transplantation for tetratricopeptide repeat domain 7A deficiency: Long-term follow-up. Blood 2016, 128, 1306–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilroy, R.K.; Coccia, P.F.; Talmadge, J.E.; Hatcher, L.I.; Pirruccello, S.J.; Shaw, B.W., Jr.; Rubocki, R.J.; Sudan, D.L.; Langnas, A.N.; Horslen, S.P. Donor immune reconstitution after liver-small bowel transplantation for multiple intestinal atresia with immunodeficiency. Blood 2004, 103, 1171–1174. [Google Scholar] [CrossRef] [PubMed]
- White, R.A.; McNulty, S.G.; Nsumu, N.N.; Boydston, L.A.; Brewer, B.P.; Shimizu, K. Positional cloning of the Ttc7 gene required for normal iron homeostasis and mutated in hea and fsn anemia mice. Genomics 2005, 85, 330–337. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Patient n.^ [Ref.° n.] | Age, Gender | TTC7A Mutation(s) (c.DNA, Protein) | Dermatological Features | Histopathology |
|---|---|---|---|---|
| 3 years, female | c.1008C>G, p.Tyr336*; c.1479delG, p.Leu493fs*13 | Diffuse xerosis | Ichthyotic epidermis, epidermal hyperplasia, follicular keratin plugs |
| 6 years, male | c.2496_2497delCG, p.A832fs*1 | Diffuse xerosis, PPK”, acral and subungual hyperkeratosis | Ichthyotic epidermis, epidermal hyperplasia, follicular keratin plugs |
| 3–7 (single-family) [5,7] | 4–50 years, 3 females, 2 males | c.211G>A, p.E71K | Progressive alopecia (all cases), toenail subungual hyperkeratosis (4 cases), psoriasiform lesions (1 case) | Not available |
| 8 [8] | 5 years, female | c.2170C>A, p.Q724K | Diffuse xerosis, eczematous lesions, and excoriations | Hyperorthokeratosis, spongiosis, eosinophilic infiltrate |
| Present case | 20 months, male | c.295A>G, p.M99V | Diffuse xerosis, follicular hyperkeratosis, mild subungual hyperkeratosis | Epidermal hyperplasia, follicular keratin plugs, apoptotic keratinocytes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diociaiuti, A.; Caruso, R.; Ricci, S.; De Vito, R.; Strocchio, L.; Castiglia, D.; Zambruno, G.; El Hachem, M. Prominent Follicular Keratosis in Multiple Intestinal Atresia with Combined Immune Deficiency Caused by a TTC7A Homozygous Mutation. Genes 2022, 13, 821. https://doi.org/10.3390/genes13050821
Diociaiuti A, Caruso R, Ricci S, De Vito R, Strocchio L, Castiglia D, Zambruno G, El Hachem M. Prominent Follicular Keratosis in Multiple Intestinal Atresia with Combined Immune Deficiency Caused by a TTC7A Homozygous Mutation. Genes. 2022; 13(5):821. https://doi.org/10.3390/genes13050821
Chicago/Turabian StyleDiociaiuti, Andrea, Roberta Caruso, Silvia Ricci, Rita De Vito, Luisa Strocchio, Daniele Castiglia, Giovanna Zambruno, and May El Hachem. 2022. "Prominent Follicular Keratosis in Multiple Intestinal Atresia with Combined Immune Deficiency Caused by a TTC7A Homozygous Mutation" Genes 13, no. 5: 821. https://doi.org/10.3390/genes13050821
APA StyleDiociaiuti, A., Caruso, R., Ricci, S., De Vito, R., Strocchio, L., Castiglia, D., Zambruno, G., & El Hachem, M. (2022). Prominent Follicular Keratosis in Multiple Intestinal Atresia with Combined Immune Deficiency Caused by a TTC7A Homozygous Mutation. Genes, 13(5), 821. https://doi.org/10.3390/genes13050821