
Development and Application of a Target Capture Sequencing SNP-Genotyping Platform in Rice

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Target Capture Panel Design and Next-generation Sequencing
2.3. Variety Diversity Analysis and QTL Mapping
3. Results
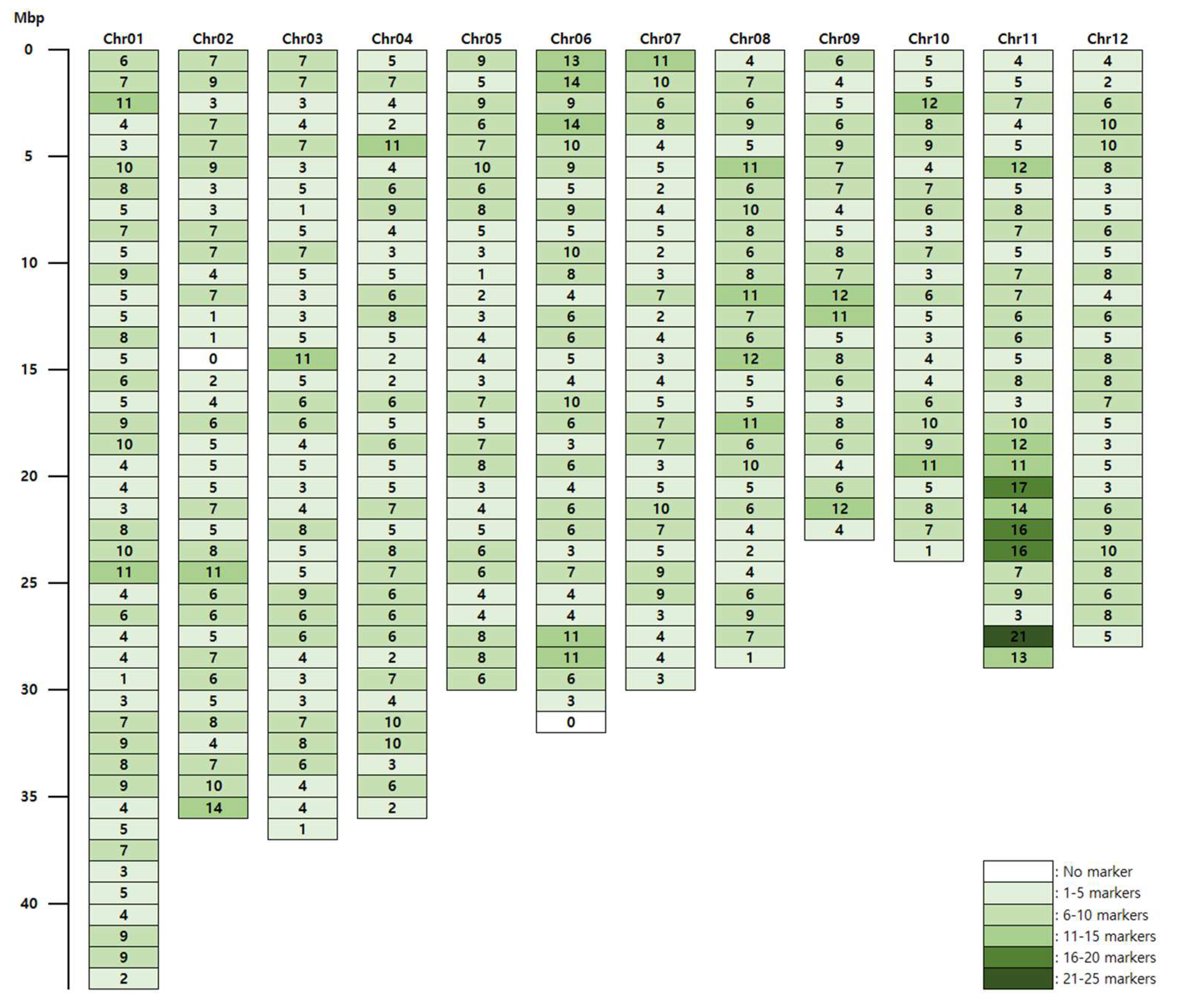
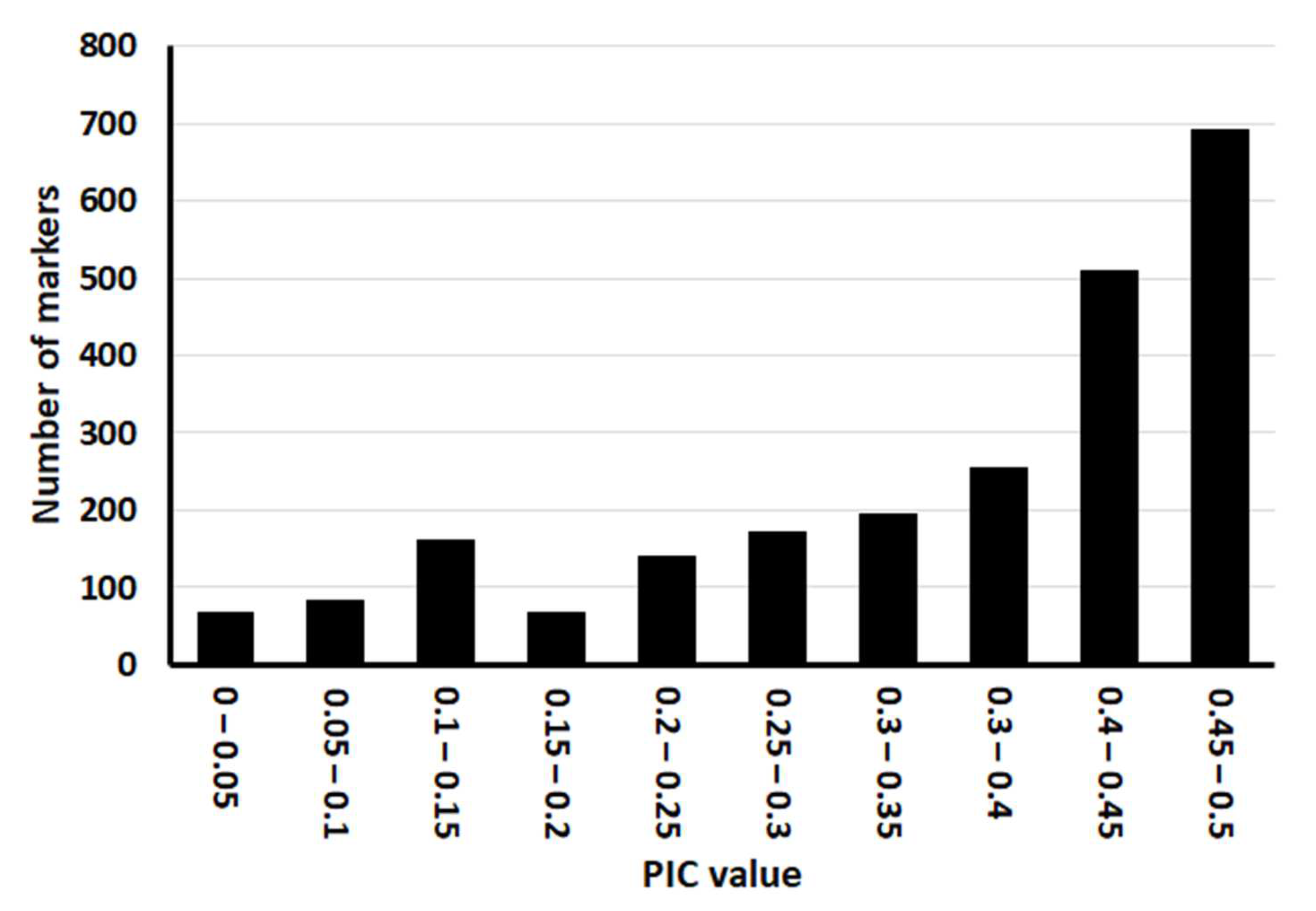
3.1. Target Capture Panel Design
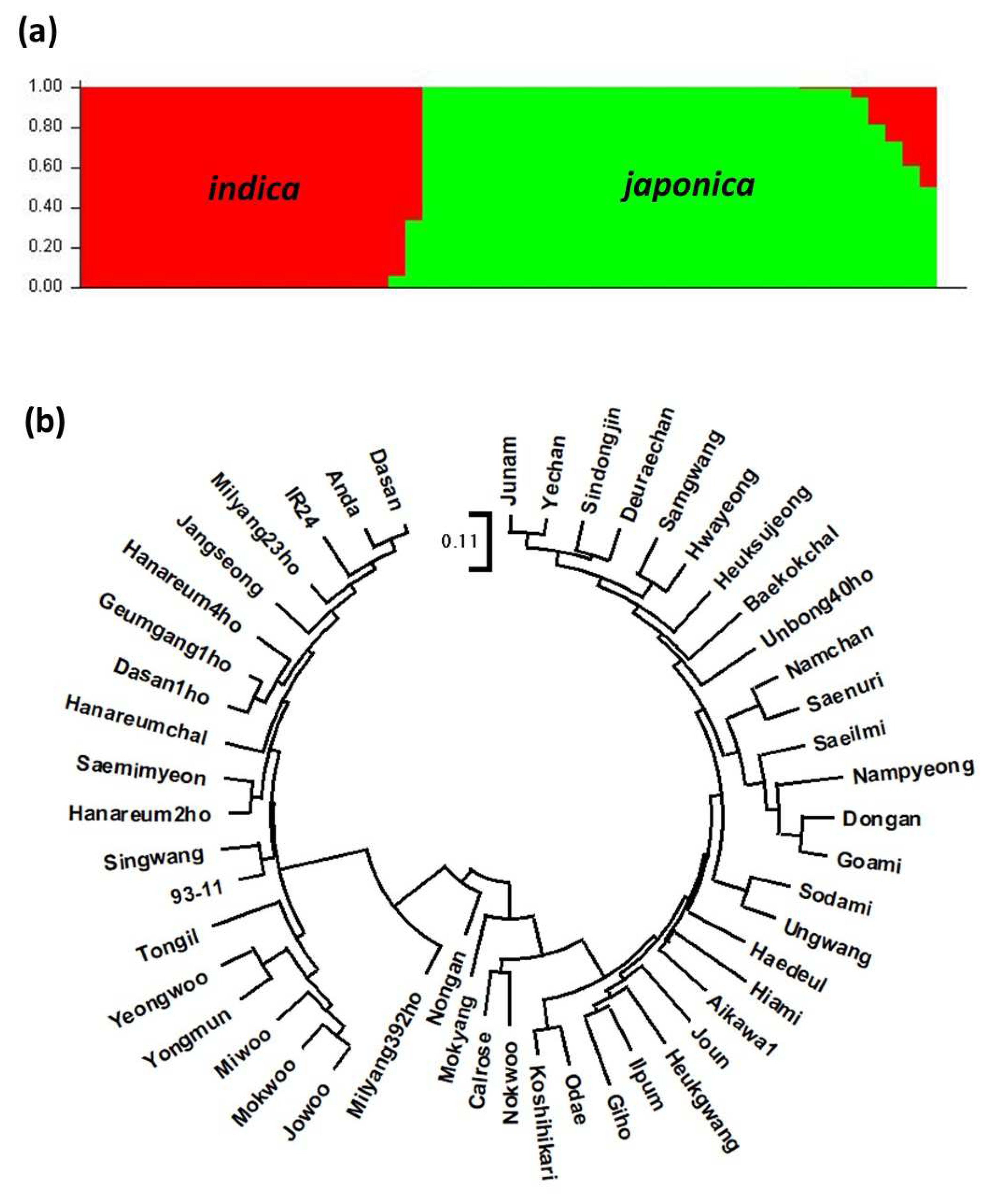
3.2. Diversity Analysis of Rice Varieties Using the Target Capture Panel
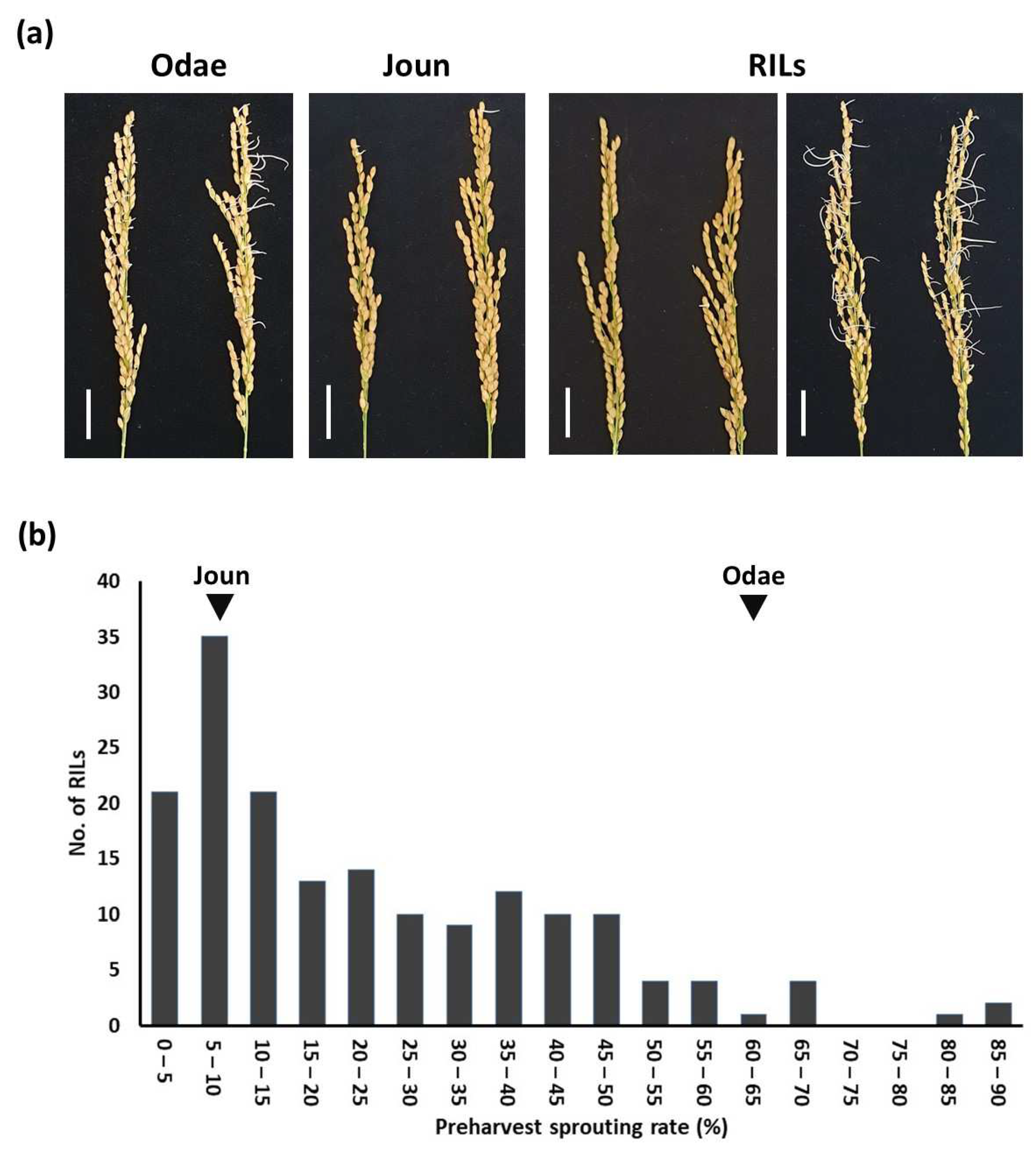
3.3. QTL Analysis of Preharvest Sprouting Resistance Using the Target Capture Panel
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wing, R.A.; Purugganan, M.D.; Zhang, Q. The rice genome revolution: From an ancient grain to Green Super Rice. Nat. Rev. Genet. 2018, 19, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xie, W.; He, H.; Yu, H.; Chen, W.; Li, J.; Yu, R.; Yao, Y.; Zhang, W.; He, Y.; et al. A High-Density SNP Genotyping Array for Rice Biology and Molecular Breeding. Mol. Plant 2014, 7, 541–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, K.; Wright, M.; Kimball, J.; Eizenga, G.; McClung, A.; Kovach, M.; Tyagi, W.; Ali, L.; Tung, C.-W.; Reynolds, A.; et al. Genomic Diversity and Introgression in O. sativa Reveal the Impact of Domestication and Breeding on the Rice Genome. PLoS ONE 2010, 5, e10780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, M.J.; Zhao, K.; Wright, M.; McNally, K.L.; Rey, J.; Tung, C.-W.; Reynolds, A.; Scheffler, B.; Eizenga, G.; McClung, A.; et al. High-throughput single nucleotide polymorphism genotyping for breeding applications in rice using the BeadXpress platform. Mol. Breed. 2011, 29, 875–886. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nagasaki, H.; Yonemaru, J.-I.; Ebana, K.; Nakajima, M.; Shibaya, T.; Yano, M. Fine definition of the pedigree haplotypes of closely related rice cultivars by means of genome-wide discovery of single-nucleotide polymorphisms. BMC Genom. 2010, 11, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Xie, W.; Li, J.; Zhou, F.; Zhang, Q. A whole-genome SNP array (RICE 6 K) for genomic breeding in rice. Plant Biotechnol. J. 2014, 12, 28–37. [Google Scholar] [CrossRef]
- Thomson, M.J.; Singh, N.; Dwiyanti, M.S.; Wang, D.R.; Wright, M.H.; Perez, F.A.; Declerck, G.; Chin, J.H.; Malitic-Layaoen, G.A.; Juanillas, V.M.; et al. Large-scale deployment of a rice 6 K SNP array for genetics and breeding applications. Rice 2017, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Morales, K.Y.; Singh, N.; Perez, F.A.; Ignacio, J.C.; Thapa, R.; Arbelaez, J.D.; Tabien, R.E.; Famoso, A.; Wang, D.R.; Septiningsih, E.M.; et al. An improved 7K SNP array, the C7AIR, provides a wealth of validated SNP markers for rice breeding and genetics studies. PLoS ONE 2020, 15, e0232479. [Google Scholar] [CrossRef]
- Zhao, K.; Tung, C.-W.; Eizenga, G.C.; Wright, M.; Ali, M.L.; Price, A.H.; Norton, G.; Islam, M.R.; Reynolds, A.R.; Mezey, J.G.; et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2011, 2, 467. [Google Scholar] [CrossRef]
- Singh, N.; Jayaswal, P.K.; Panda, K.; Mandal, P.; Kumar, V.; Singh, B.; Mishra, S.; Singh, Y.; Singh, R.; Rai, V.; et al. Single-copy gene based 50 K SNP chip for genetic studies and molecular breeding in rice. Sci. Rep. 2015, 5, 11600. [Google Scholar] [CrossRef]
- McCouch, S.R.; Wright, M.; Tung, C.-W.; Maron, L.; McNally, K.; Fitzgerald, M.; Singh, N.; Declerck, G.; Agosto-Perez, F.; Korniliev, P.; et al. Open access resources for genome-wide association mapping in rice. Nat. Commun. 2016, 7, 10532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, M.J. High-Throughput SNP Genotyping to Accelerate Crop Improvement. Plant Breed. Biotechnol. 2014, 2, 195–212. [Google Scholar] [CrossRef]
- Arbelaez, J.D.; Dwiyanti, M.S.; Tandayu, E.; Llantada, K.; Jarana, A.; Ignacio, J.C.; Platten, J.D.; Cobb, J.; Rutkoski, J.E.; Thomson, M.J.; et al. 1k-RiCA (1K-Rice Custom Amplicon) a novel genotyping amplicon-based SNP assay for genetics and breeding applications in rice. Rice 2019, 12, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Wang, H.; Tao, J.; Ren, Y.; Xu, C.; Wu, K.; Zou, C.; Zhang, J.; Xu, Y. Development of multiple SNP marker panels affordable to breeders through genotyping by target sequencing (GBTS) in maize. Mol. Breed. 2019, 39, 37. [Google Scholar] [CrossRef]
- Guo, Z.; Yang, Q.; Huang, F.; Zheng, H.; Sang, Z.; Xu, Y.; Zhang, C.; Wu, K.; Tao, J.; Prasanna, B.M.; et al. Development of high-resolution multiple-SNP arrays for genetic analyses and molecular breeding through genotyping by target sequencing and liquid chip. Plant Commun. 2021, 2, 100230. [Google Scholar] [CrossRef]
- Wang, C.; Liu, R.; Liu, Y.; Hou, W.; Wang, X.; Miao, Y.; He, Y.; Ma, Y.; Li, G.; Wang, D.; et al. Development and application of the Faba_bean_130K targeted next-generation sequencing SNP genotyping platform based on transcriptome sequencing. Theor. Appl. Genet. 2021, 134, 3195–3207. [Google Scholar] [CrossRef]
- Pootakham, W.; Shearman, J.R.; Ruang-Areerate, P.; Sonthirod, C.; Sangsrakru, D.; Jomchai, N.; Yoocha, T.; Triwitayakorn, K.; Tragoonrung, S.; Tangphatsornruang, S. Large-Scale SNP Discovery through RNA Sequencing and SNP Genotyping by Targeted Enrichment Sequencing in Cassava (Manihot esculenta Crantz). PLoS ONE 2014, 9, e116028. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Chen, S.; Chen, L.; Sun, K.; Huang, C.; Zhou, D.; Huang, Y.; Wang, J.; Liu, Y.; Wang, H.; et al. Development of a core SNP arrays based on the KASP method for molecular breeding of rice. Rice 2019, 12, 21. [Google Scholar] [CrossRef]
- Cheon, K.-S.; Baek, J.; Cho, Y.-I.; Jeong, Y.-M.; Lee, Y.-Y.; Oh, J.; Won, Y.J.; Kang, D.-Y.; Oh, H.; Kim, S.L.; et al. Single Nucleotide Polymorphism (SNP) Discovery and Kompetitive Allele-Specific PCR (KASP) Marker Development with Korean Japonica Rice Varieties. Plant Breed. Biotechnol. 2018, 6, 391–403. [Google Scholar] [CrossRef]
- Cheon, K.-S.; Jeong, Y.-M.; Lee, Y.-Y.; Oh, J.; Kang, D.-Y.; Oh, H.; Kim, S.L.; Kim, N.; Lee, E.; Baek, J.; et al. Kompetitive Allele-Specific PCR Marker Development and Quantitative Trait Locus Mapping for Bakanae Disease Resistance in Korean Japonica Rice Varieties. Plant Breed. Biotechnol. 2019, 7, 208–219. [Google Scholar] [CrossRef]
- Cheon, K.-S.; Jeong, Y.-M.; Oh, H.; Oh, J.; Kang, D.-Y.; Kim, N.; Lee, E.; Baek, J.; Kim, S.L.; Choi, I.; et al. Development of 454 New Kompetitive Allele-Specific PCR (KASP) Markers for Temperate japonica Rice Varieties. Plants 2020, 9, 1531. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.-Y.; Cheon, K.-S.; Oh, J.; Oh, H.; Kim, S.L.; Kim, N.; Lee, E.; Choi, I.; Baek, J.; Kim, K.-H.; et al. Rice Genome Resequencing Reveals a Major Quantitative Trait Locus for Resistance to Bakanae Disease Caused by Fusarium fujikuroi. Int. J. Mol. Sci. 2019, 20, 2598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheon, K.-S.; Won, Y.J.; Jeong, Y.-M.; Lee, Y.-Y.; Kang, D.-Y.; Oh, J.; Oh, H.; Kim, S.L.; Kim, N.; Lee, E.; et al. QTL mapping for pre-harvest sprouting resistance in japonica rice varieties utilizing genome re-sequencing. Mol. Genet. Genom. 2020, 295, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Won, Y.J.; Lee, C.; Cheon, K.-S.; Oh, H.; Lee, G.-S.; Baek, J.; Yoon, I.S.; Kim, S.L.; Cha, Y.-S.; et al. Identification of Grain Size-Related QTLs in Korean japonica Rice Using Genome Resequencing and High-Throughput Image Analysis. Agriculture 2022, 12, 51. [Google Scholar] [CrossRef]
- Lee, C.-M.; Suh, J.-P.; Park, H.-S.; Baek, M.-K.; Jeong, O.-Y.; Yun, S.-J.; Cho, Y.-C.; Kim, S.-M. Identification of QTL Combinations that Cause Spikelet Sterility in Rice Derived from Interspecific Crosses. Rice 2021, 14, 99. [Google Scholar] [CrossRef]
- Kang, J.-W.; Shin, D.; Cho, J.-H.; Lee, J.-Y.; Kwon, Y.; Park, D.-S.; Ko, J.-M.; Lee, J.-H. Accelerated development of rice stripe virus-resistant, near-isogenic rice lines through marker-assisted backcrossing. PLoS ONE 2019, 14, e0225974. [Google Scholar] [CrossRef]
- Kim, M.-S.; Yang, J.-Y.; Yu, J.-K.; Lee, Y.; Park, Y.-J.; Kang, K.-K.; Cho, Y.-G. Breeding of High Cooking and Eating Quality in Rice by Marker-Assisted Backcrossing (MABc) Using KASP Markers. Plants 2021, 10, 804. [Google Scholar] [CrossRef]
- Kim, M.-S.; Yu, J.-K.; Ko, S.-R.; Kim, K.-J.; Ji, H.; Kang, K.-K.; Cho, Y.-G. Marker-Assisted Backcrossing (MABc) to Improve Eating Quality with Thin Seed Coat and Aleurone Layer of Non-Glutinous Japonica Variety in Rice. Genes 2022, 13, 210. [Google Scholar] [CrossRef]
- Sasaki, A.; Ashikari, M.; Ueguchi-Tanaka, M.; Itoh, H.; Nishimura, A.; Swapan, D.; Ishiyama, K.; Saito, T.; Kobayashi, M.; Khush, G.S.; et al. Green revolution: A mutant gibberellin-synthesis gene in rice. Nature 2002, 416, 701–702. [Google Scholar] [CrossRef]
- Serrote, C.M.L.; Reiniger, L.R.S.; Silva, K.B.; Rabaiolli, S.M.D.S.; Stefanel, C.M. Determining the Polymorphism Information Content of a molecular marker. Gene 2020, 726, 144175. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of Population Structure Using Multilocus Genotype Data: Linked Loci and Correlated Allele Frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, H.; Zhang, L.; Meng, L. (Eds.) Manual of QTL IciMapping; The Quantitative Genetics Group, Institute of Crop Science, Chinese Academy of Agricultural Sciences (CAAS), and Generic Resources Program; International Maize and Wheat Improvement Center (CIMMYT): Beijing, China; Mexico City, Mexico, 2016. [Google Scholar]
- Lee, J.-S.; Chebotarov, D.; McNally, K.L.; Pede, V.; Setiyono, T.D.; Raquid, R.; Hyun, W.-J.; Jeung, J.-U.; Kohli, A.; Mo, Y. Novel Sources of Pre-Harvest Sprouting Resistance for Japonica Rice Improvement. Plants 2021, 10, 1709. [Google Scholar] [CrossRef]
- Zhu, D.; Qian, Z.; Wei, H.; Guo, B.; Xu, K.; Dai, Q.; Zhang, H.; Huo, Z. The effects of field pre-harvest sprouting on the morphological structure and physicochemical properties of rice (Oryza sativa L.) starch. Food Chem. 2019, 278, 10–16. [Google Scholar] [CrossRef]
- Sohn, S.-I.; Pandian, S.; Kumar, T.S.; Zoclanclounon, Y.A.B.; Muthuramalingam, P.; Shilpha, J.; Satish, L.; Ramesh, M. Seed Dormancy and Pre-Harvest Sprouting in Rice—An Updated Overview. Int. J. Mol. Sci. 2021, 22, 11804. [Google Scholar] [CrossRef]
- Zhou, Y.; Xie, Y.; Cai, J.; Liu, C.; Zhu, H.; Jiang, R.; Zhong, Y.; Zhang, G.; Tan, B.; Liu, G.; et al. Substitution mapping of QTLs controlling seed dormancy using single segment substitution lines derived from multiple cultivated rice donors in seven cropping seasons. Theor. Appl. Genet. 2017, 130, 1191–1205. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Mean | Minimum | Maximum |
|---|---|---|---|
| All varieties | 854 | 159 | 1368 |
| Within japonica | 623 | 159 | 916 |
| Within indica | 491 | 162 | 861 |
| Between japonica and indica | 1177 | 603 | 1368 |
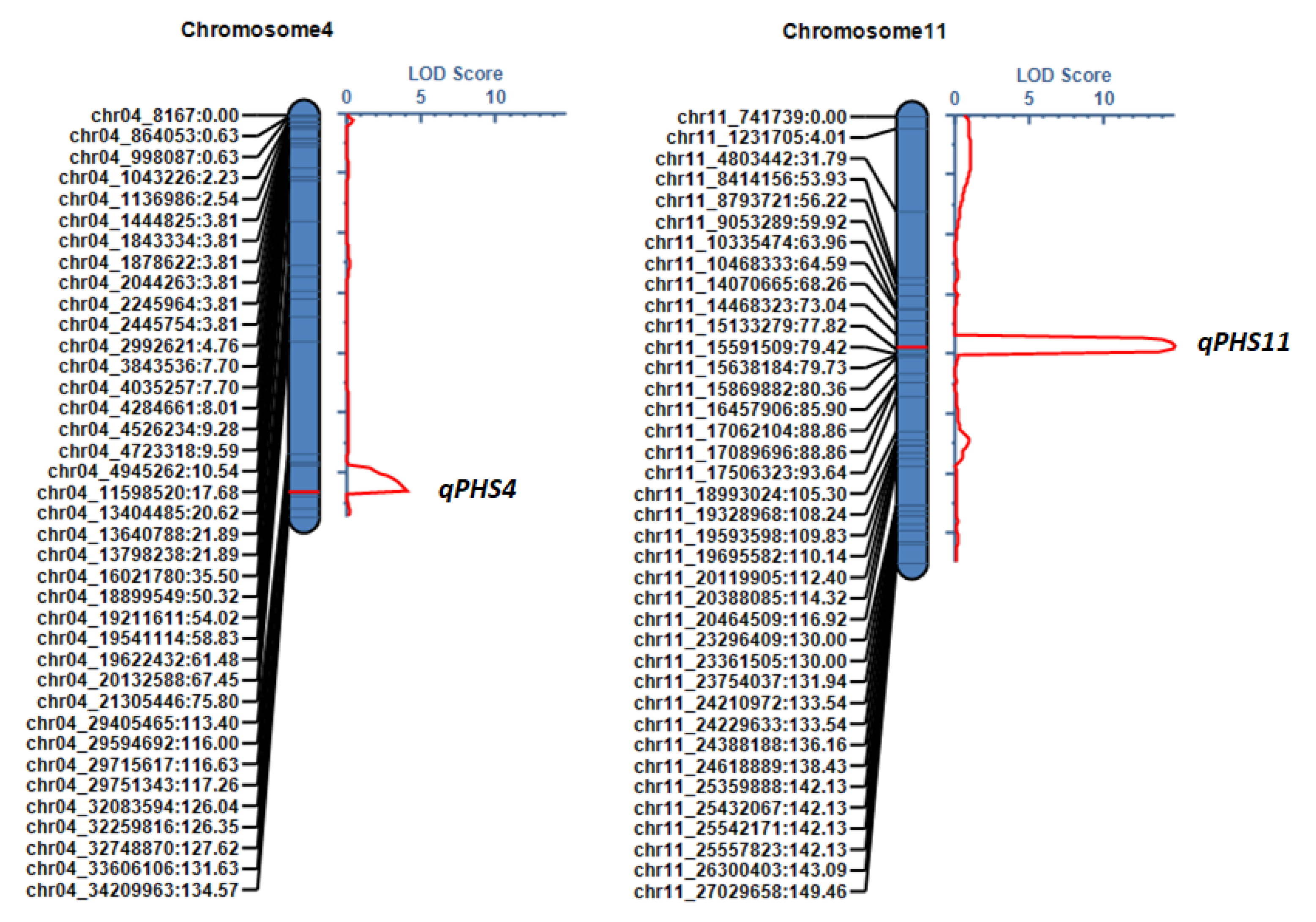
| QTL name | Chromosome | Location (cM) | QTL Interval * (cM) | LOD | Additive Effect | PVE (%) ** |
|---|---|---|---|---|---|---|
| qPHS4 | 4 | 126 | 121.5–126.5 | 4.06 | −4.84 | 6.48 |
| qPHS11 | 11 | 77 | 74.5–78.5 | 14.74 | −10.13 | 28.66 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.; Cheon, K.-S.; Shin, Y.; Oh, H.; Jeong, Y.-M.; Jang, H.; Park, Y.-C.; Kim, K.-Y.; Cho, H.-C.; Won, Y.-J.; et al. Development and Application of a Target Capture Sequencing SNP-Genotyping Platform in Rice. Genes 2022, 13, 794. https://doi.org/10.3390/genes13050794
Lee C, Cheon K-S, Shin Y, Oh H, Jeong Y-M, Jang H, Park Y-C, Kim K-Y, Cho H-C, Won Y-J, et al. Development and Application of a Target Capture Sequencing SNP-Genotyping Platform in Rice. Genes. 2022; 13(5):794. https://doi.org/10.3390/genes13050794
Chicago/Turabian StyleLee, Chaewon, Kyeong-Seong Cheon, Yunji Shin, Hyoja Oh, Young-Min Jeong, Hoon Jang, Yong-Chan Park, Kyung-Yun Kim, Hang-Chul Cho, Yong-Jae Won, and et al. 2022. "Development and Application of a Target Capture Sequencing SNP-Genotyping Platform in Rice" Genes 13, no. 5: 794. https://doi.org/10.3390/genes13050794
APA StyleLee, C., Cheon, K.-S., Shin, Y., Oh, H., Jeong, Y.-M., Jang, H., Park, Y.-C., Kim, K.-Y., Cho, H.-C., Won, Y.-J., Baek, J., Cha, Y.-S., Kim, S.-L., Kim, K.-H., & Ji, H. (2022). Development and Application of a Target Capture Sequencing SNP-Genotyping Platform in Rice. Genes, 13(5), 794. https://doi.org/10.3390/genes13050794