
Sensitive and Specific Detection of Lumpy Skin Disease Virus in Cattle by CRISPR-Cas12a Fluorescent Assay Coupled with Recombinase Polymerase Amplification

 ,
,  and
and 
Abstract
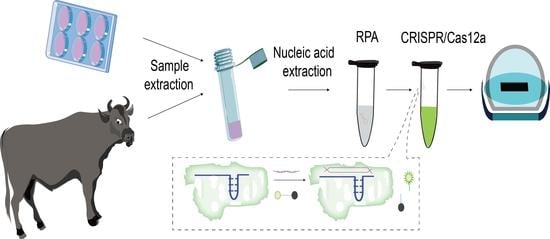
1. Introduction
2. Materials and Methods
2.1. Primer Design and Polymerase Chain Reaction
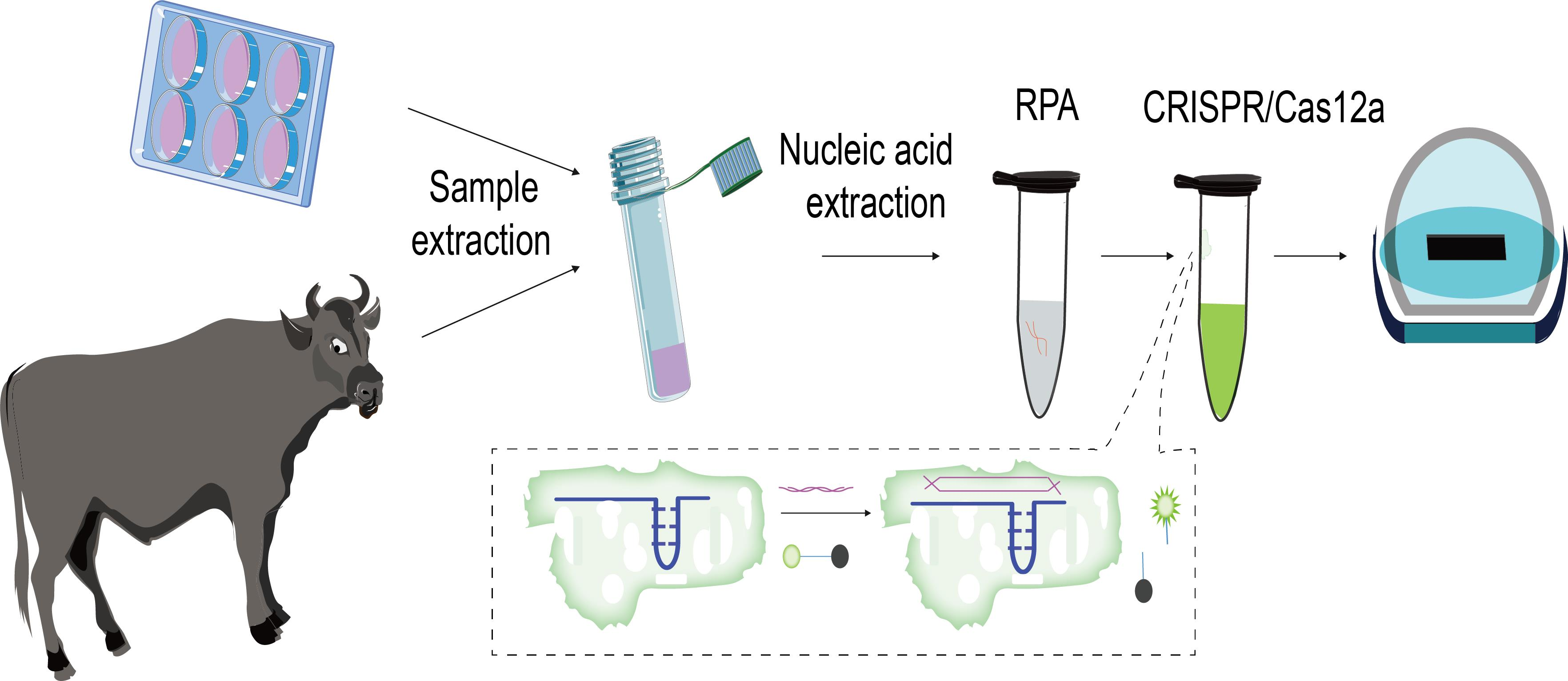
2.2. Preparation of CRISPR RNA (crRNAs) Targeting LSDV orf068
2.3. PCR or Quantitative PCR (qPCR) Coupled with CRISPR-Cas12a Assay
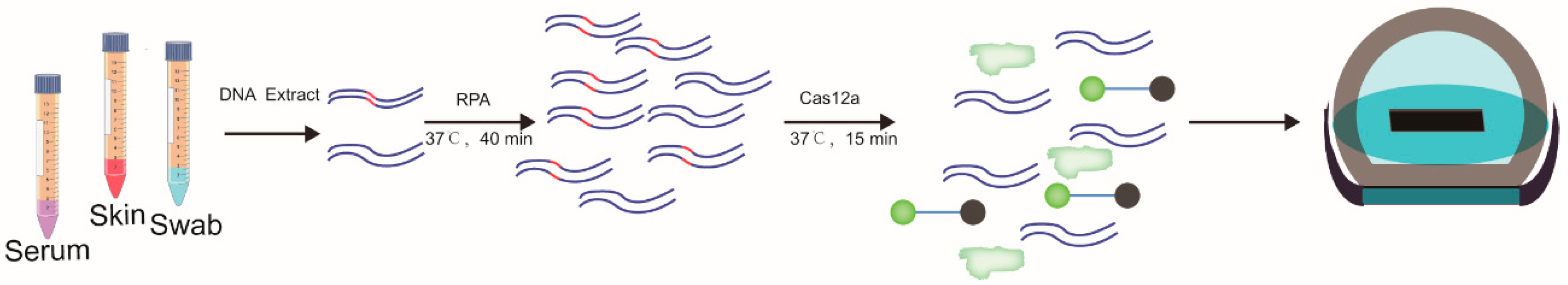
2.4. Recombinase Polymerase Amplification (RPA) Coupled with CRISPR-Cas12a Assay
2.5. LSDV Detection Sensitivity by RPA-Cas12a-Fluorescence Assay
2.6. LSDV Detection Specificity by RPA-Cas12a-Fluorescence Assay
2.7. Diagnosis of Lumpy Skin Disease by RPA-Cas12a-Fluorescence Assay
2.8. Statistical Analysis
3. Results
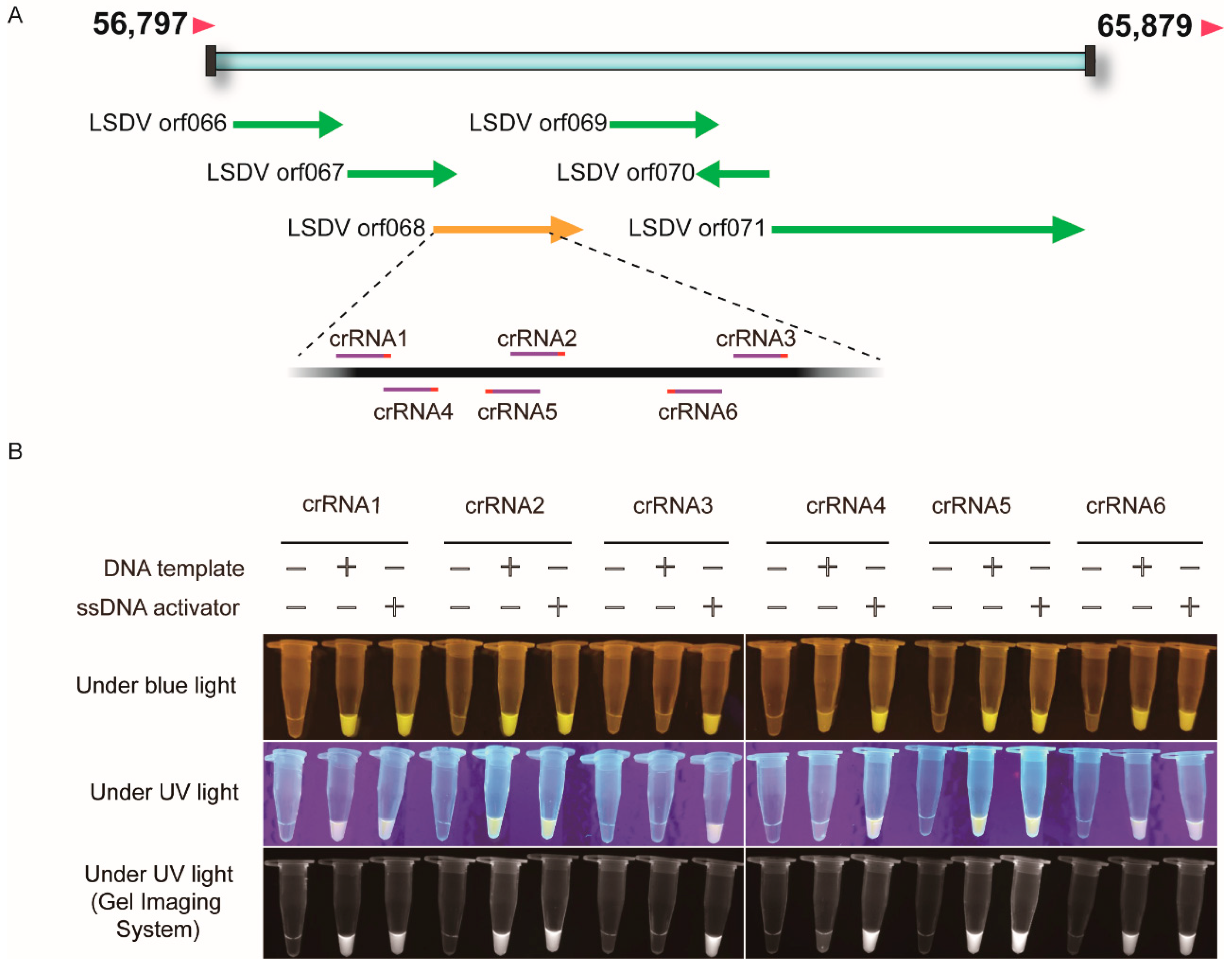
3.1. Establishment of Cas12a-Fluorescence Assay for Detection of LSDV
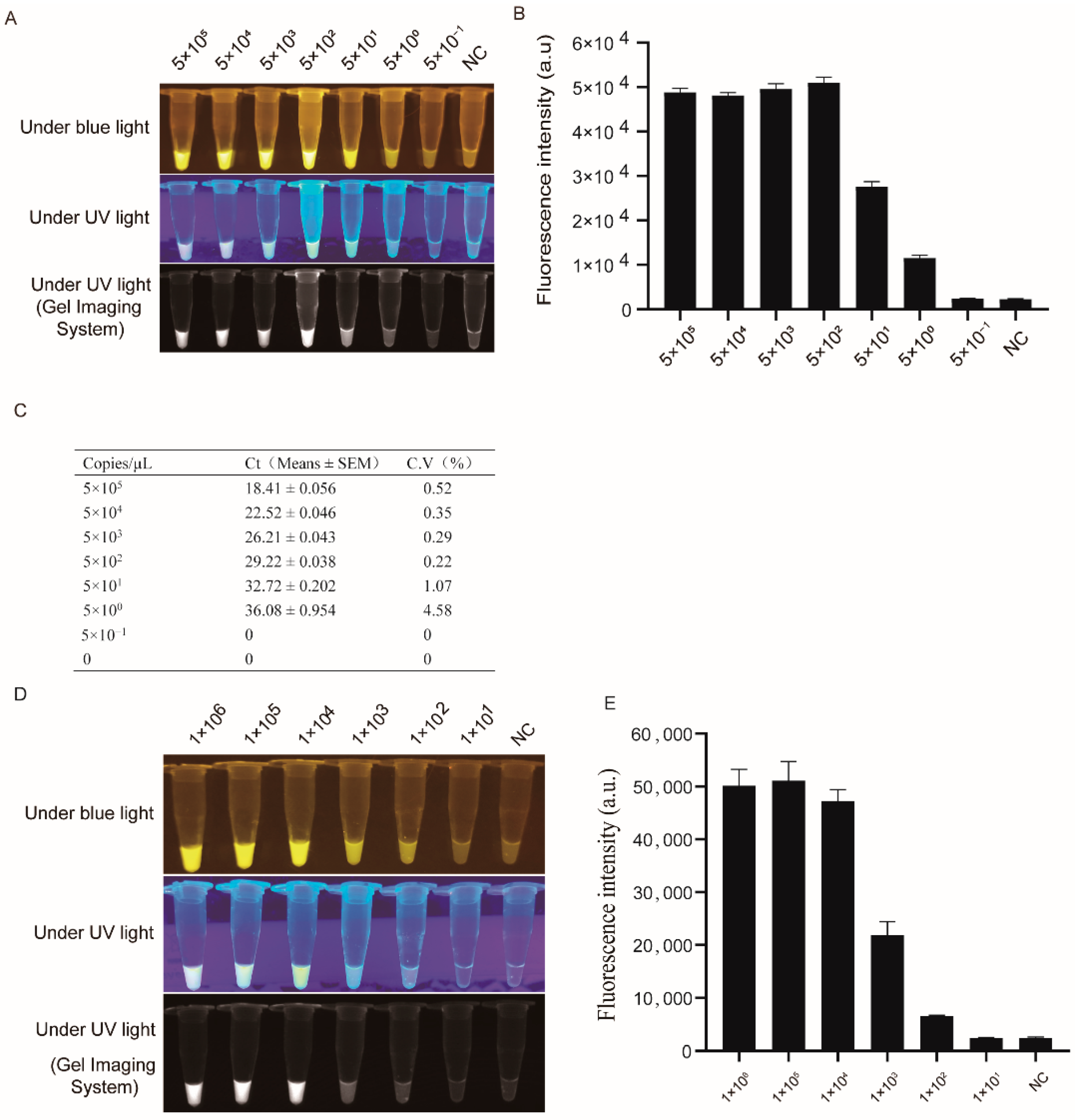
3.2. LSDV Detection Sensitivity by RPA-Cas12a-Fluorescence Assay
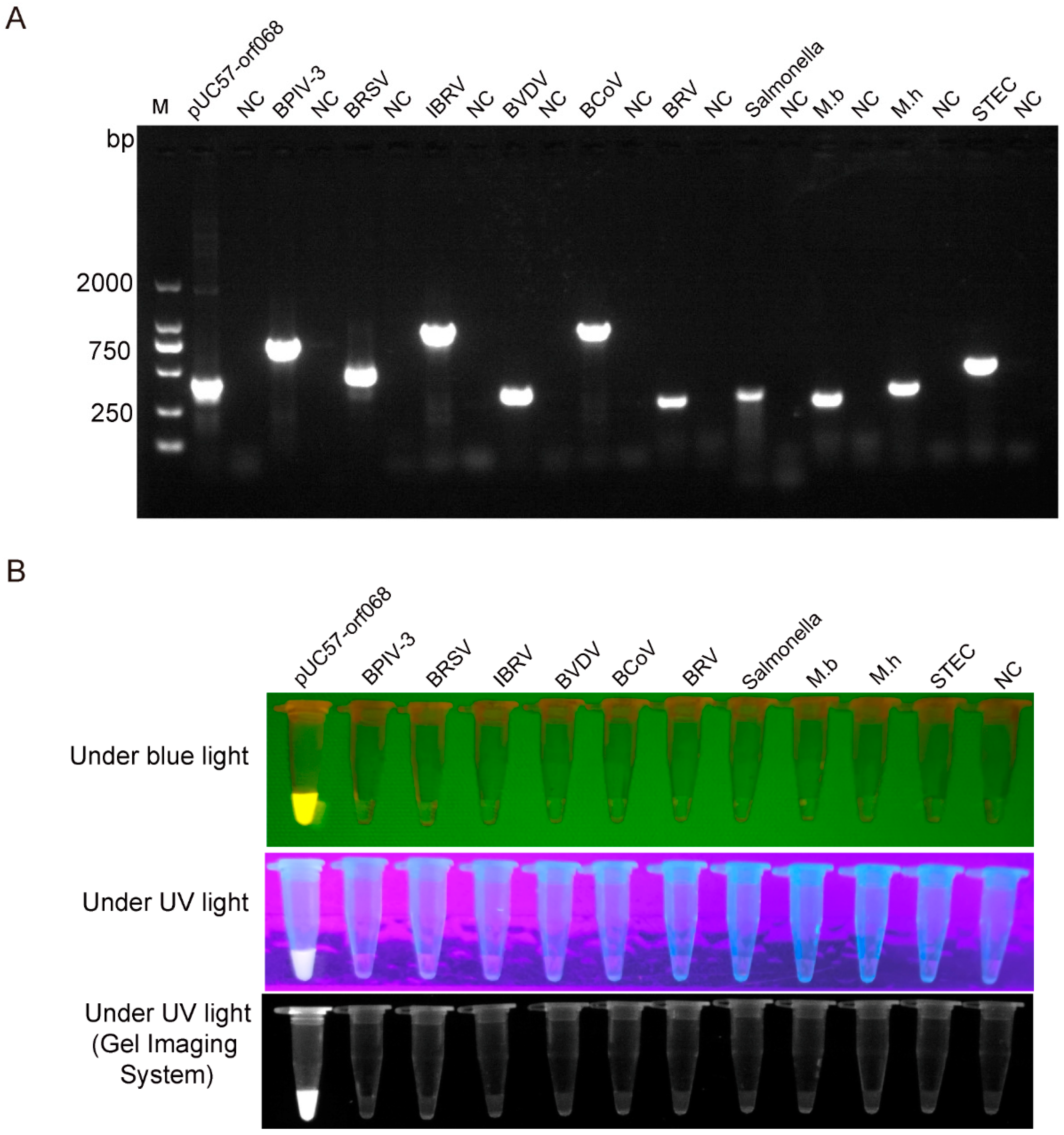
3.3. LSDV Detection Specificity by RPA-Cas12a-Fluorescence Assay
3.4. Detection of LSDV in Clinical Samples by RPA-Cas12a-Fluorescence Assay
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lamien, C.E.; Lelenta, M.; Goger, W.; Silber, R.; Tuppurainen, E.; Matijevic, M.; Luckins, A.G.; Diallo, A. Real time PCR method for simultaneous detection, quantitation and differentiation of capripoxviruses. J. Virol. Methods 2011, 171, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Tulman, E.R.; Afonso, C.L.; Lu, Z.; Zsak, L.; Kutish, G.F.; Rock, L.D. Genome of lumpy skin disease virus. J. Virol. 2001, 75, 7122–7130. [Google Scholar] [CrossRef] [PubMed]
- Tuppurainen, E.S.; Venter, E.H.; Coetzer, J.A. The detection of lumpy skin disease virus in samples of experimentally infected cattle using different diagnostic techniques. Onderstepoort. J. Veter. Res. 2005, 72, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Carn, V.M.; Kitching, R.P. The clinical response of cattle experimentally infected with lumpy skin disease (Neethling) virus. Arch. Virol. 1995, 140, 503–513. [Google Scholar] [CrossRef]
- Babiuk, S.; Bowden, T.R.; Boyle, D.B.; Wallace, D.B.; Kitching, R.P. Capripoxviruses: An emerging worldwide threat to sheep, goats and cattle. Transbound. Emerg. Dis. 2008, 55, 263–272. [Google Scholar] [CrossRef]
- Zhang, M.; Sun, Y.; Liu, W.; Liu, R.; Wang, X.; Bu, Z. Isolation and identification of lumpy skin disease virus from the first outbreak in China. Chin. J. Prev. Vet. Med. 2020, 42, 1058–1061. [Google Scholar]
- Sanz-Bernardo, B.; Haga, I.R.; Wijesiriwardana, N.; Basu, S.; Larner, W.; Diaz, A.V.; Langlands, Z.; Denison, E.; Stoner, J.; White, M.; et al. Quantifying and Modeling the Acquisition and Retention of Lumpy Skin Disease Virus by Hematophagus Insects Reveals Clinically but Not Subclinically Affected Cattle Are Promoters of Viral Transmission and Key Targets for Control of Disease Outbreaks. J. Virol. 2021, 95, e02239-20. [Google Scholar] [CrossRef]
- Manual of Diagnostic Tests and Vaccines for Terrestrial Animals. Available online: https://www.oie.int/en/home/ (accessed on 10 January 2022).
- Bowden, T.R.; Coupar, B.E.; Babiuk, S.L.; White, J.R.; Boyd, V.; Duch, C.J.; Shiell, B.J.; Ueda, N.; Parkyn, G.R.; Copps, J.S.; et al. Detection of antibodies specific for sheeppox and goatpox viruses using recombinant capripoxvirus antigens in an indirect enzyme-linked immunosorbent assay. J. Virol. Methods 2009, 161, 19–29. [Google Scholar] [CrossRef]
- Heine, H.; Stevens, M.; Foord, A.; Boyle, D. A capripoxvirus detection PCR and antibody ELISA based on the major antigen P32, the homolog of the vaccinia virus H3L gene. J. Immunol. Methods 1999, 227, 187–196. [Google Scholar] [CrossRef]
- Gari, G.; Biteau-Coroller, F.; LeGoff, C.; Caufour, P.; Roger, F. Evaluation of indirect fluorescent antibody test (IFAT) for the di-agnosis and screening of lumpy skin disease using Bayesian method. Vet. Microbiol. 2008, 129, 269–280. [Google Scholar] [CrossRef]
- Carn, V.; Kitching, R.; Hammond, J.; Chand, P. Use of a recombinant antigen in an indirect ELISA for detecting bovine antibody to capripoxvirus. J. Virol. Methods 1994, 49, 285–294. [Google Scholar] [CrossRef]
- Kitching, R.P.; McGrane, J.J.; Taylor, W.P. Capripox in the Yemen Arab Republic and the Sultanate of Oman. Trop. Anim. Health Prod. 1986, 18, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Ireland, D.; Binepal, Y. Improved detection of capripoxvirus in biopsy samples by PCR. J. Virol. Methods 1998, 74, 1–7. [Google Scholar] [CrossRef]
- Das, A.; Deng, M.Y.; Babiuk, S.; McIntosh, M.T. Modification of two capripoxvirus quantitative real-time PCR assays to improve diagnostic sensitivity and include beta-actin as an internal positive control. J. Veter. Diagn. Investig. 2017, 29, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Soroka, M.; Wasowicz, B.; Rymaszewska, A. Loop-Mediated Isothermal Amplification (LAMP): The Better Sibling of PCR? Cells 2021, 10, 1931. [Google Scholar] [CrossRef]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef]
- Tao, D.; Liu, J.; Nie, X.; Xu, B.; Tran-Thi, T.-N.; Niu, L.; Liu, X.; Ruan, J.; Lan, X.; Peng, G.; et al. Application of CRISPR-Cas12a Enhanced Fluorescence Assay Coupled with Nucleic Acid Amplification for the Sensitive Detection of African Swine Fever Virus. ACS Synth. Biol. 2020, 9, 2339–2350. [Google Scholar] [CrossRef]
- Yang, B.; Shi, Z.; Ma, Y.; Wang, L.; Cao, L.; Luo, J.; Wan, Y.; Song, R.; Yan, Y.; Yuan, K.; et al. LAMP assay coupled with CRISPR/Cas12a system for portable detection of African swine fever virus. Transbound. Emerg. Dis. 2021, 1–8. [Google Scholar] [CrossRef]
- Ren, M.; Mei, H.; Zhou, M.; Fu, Z.F.; Han, H.; Bi, D.; Peng, F.; Zhao, L. Development of A Super-Sensitive Diagnostic Method for African Swine Fever Using CRISPR Techniques. Virol. Sin. 2021, 36, 220–230. [Google Scholar] [CrossRef]
- Wei, N.; Zheng, B.; Niu, J.; Chen, T.; Ye, J.; Si, Y.; Cao, S. Rapid Detection of Genotype II African Swine Fever Virus Using CRISPR Cas13a-Based Lateral Flow Strip. Viruses 2022, 14, 179. [Google Scholar] [CrossRef]
- Broughton, J.P.; Deng, X.; Yu, G.; Fasching, C.L.; Servellita, V.; Singh, J.; Miao, X.; Streithorst, J.A.; Granados, A.; So-tomayor-Gonzalez, A.; et al. CRISPR-Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020, 38, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Yin, K.; Li, Z.; Lalla, R.V.; Ballesteros, E.; Sfeir, M.M.; Liu, C. Ultrasensitive and visual detection of SARS-CoV-2 using all-in-one dual CRISPR-Cas12a assay. Nat. Commun. 2020, 11, 4711. [Google Scholar] [CrossRef] [PubMed]
- Kellner, M.J.; Koob, J.G.; Gootenberg, J.S.; Abudayyeh, O.O.; Zhang, F. SHERLOCK: Nucleic acid detection with CRISPR nucleases. Nat. Protoc. 2019, 14, 2986–3012. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Xu, J.; Liu, Y.; Peng, H.; Feng, W.; Cao, Y.; Wu, J.; Xiao, H.; Pabbaraju, K.; Tipples, G.; et al. Isothermal Amplification and Ambient Visualization in a Single Tube for the Detection of SARS-CoV-2 Using Loop-Mediated Amplification and CRISPR Technology. Anal. Chem. 2020, 92, 16204–16212. [Google Scholar] [CrossRef]
- Ai, J.-W.; Zhou, X.; Xu, T.; Yang, M.; Chen, Y.; He, G.-Q.; Pan, N.; Cai, Y.; Li, Y.; Wang, X.; et al. CRISPR-based rapid and ultra-sensitive diagnostic test for Mycobacterium tuberculosis. Emerg. Microbes Infect. 2019, 8, 1361–1369. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Li, S.; Zhu, X.; Wang, X.; Huang, J.; Yang, X.; Tai, J. LAMP-CRISPR-Cas12-based diagnostic platform for detection of Mycobacterium tuberculosis complex using real-time fluorescence or lateral flow test. Mikrochim. Acta 2021, 188, 1–9. [Google Scholar] [CrossRef]
- Zhou, J.; Yin, L.; Dong, Y.; Peng, L.; Liu, G.; Man, S.; Ma, L. CRISPR-Cas13a based bacterial detection platform: Sensing pathogen Staphylococcus aureus in food samples. Anal. Chim. Acta 2020, 1127, 225–233. [Google Scholar] [CrossRef]
- Li, Y.; Deng, F.; Hall, T.; Vesey, G.; Goldys, E.M. CRISPR/Cas12a-powered immunosensor suitable for ultra-sensitive whole Cryptosporidium oocyst detection from water samples using a plate reader. Water Res. 2021, 203, 117553. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef]
- Balinsky, C.A.; Delhon, G.; Smoliga, G.; Prarat, M.; French, R.A.; Geary, S.J.; Rock, D.L.; Rodriguez, L.L. Rapid Preclinical Detection of Sheeppox Virus by a Real-Time PCR Assay. J. Clin. Microbiol. 2008, 46, 438–442. [Google Scholar] [CrossRef]
- Batra, K.; Kumar, A.; Kumar, V.; Nanda, T.; Maan, N.S.; Maan, S. Development and evaluation of loop-mediated isothermal am-plification assay for rapid detection of Capripoxvirus. Vet. World 2015, 8, 1286–1292. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, B.; Wang, R.; Wang, D.; Wu, J.; Li, J.; Wang, J.; Liu, H.; Wang, Y. Cas12aVDet: A CRISPR/Cas12a-Based Platform for Rapid and Visual Nucleic Acid Detection. Anal. Chem. 2019, 91, 12156–12161. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yu, L.; Liu, C.; Ye, S.; Chen, W.; Li, D.; Huang, W. One-tube SARS-CoV-2 detection platform based on RT-RPA and CRISPR/Cas12a. J. Transl. Med. 2021, 19, 74. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Tao, D.; Liao, Y.; Yang, Y.; Sun, S.; Zhao, Y.; Yang, P.; Tang, Y.; Chen, B.; Liu, Y.; et al. Highly Sensitive CRISPR/Cas12a-Based Fluorescence Detection of Porcine Reproductive and Respiratory Syndrome Virus. ACS Synth. Biol. 2021, 10, 2499–2507. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| qPCR Test | Total | |||
|---|---|---|---|---|
| + | − | |||
| RPA-Cas12a-fluorescence assay | + | 26 | 1 | 27 |
| − | 1 | 12 | 13 | |
| Total | 27 | 13 | 40 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, C.; Tao, D.; Geng, Y.; Yang, H.; Xu, B.; Chen, Y.; Hu, C.; Chen, H.; Xie, S.; Guo, A. Sensitive and Specific Detection of Lumpy Skin Disease Virus in Cattle by CRISPR-Cas12a Fluorescent Assay Coupled with Recombinase Polymerase Amplification. Genes 2022, 13, 734. https://doi.org/10.3390/genes13050734
Jiang C, Tao D, Geng Y, Yang H, Xu B, Chen Y, Hu C, Chen H, Xie S, Guo A. Sensitive and Specific Detection of Lumpy Skin Disease Virus in Cattle by CRISPR-Cas12a Fluorescent Assay Coupled with Recombinase Polymerase Amplification. Genes. 2022; 13(5):734. https://doi.org/10.3390/genes13050734
Chicago/Turabian StyleJiang, Chuanwen, Dagang Tao, Yuanchen Geng, Hao Yang, Bingrong Xu, Yingyu Chen, Changmin Hu, Huanchun Chen, Shengsong Xie, and Aizhen Guo. 2022. "Sensitive and Specific Detection of Lumpy Skin Disease Virus in Cattle by CRISPR-Cas12a Fluorescent Assay Coupled with Recombinase Polymerase Amplification" Genes 13, no. 5: 734. https://doi.org/10.3390/genes13050734
APA StyleJiang, C., Tao, D., Geng, Y., Yang, H., Xu, B., Chen, Y., Hu, C., Chen, H., Xie, S., & Guo, A. (2022). Sensitive and Specific Detection of Lumpy Skin Disease Virus in Cattle by CRISPR-Cas12a Fluorescent Assay Coupled with Recombinase Polymerase Amplification. Genes, 13(5), 734. https://doi.org/10.3390/genes13050734