
Severe Infantile Axonal Neuropathy with Respiratory Failure Caused by Novel Mutation in X-Linked LAS1L Gene

 , , , , and
, , , , and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Case Report
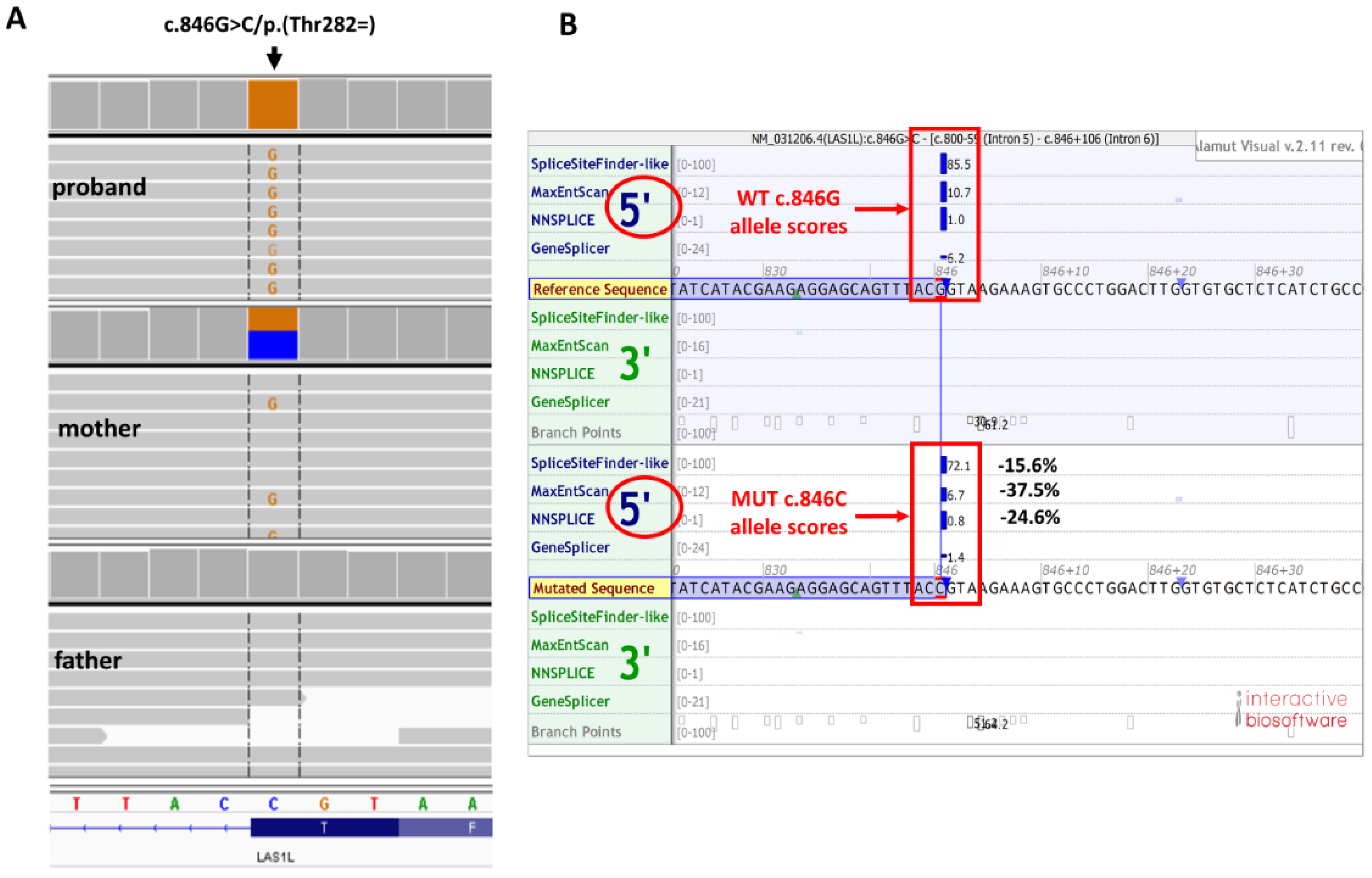
3. Genetic Testing
3.1. Whole Exome Sequencing (WES)
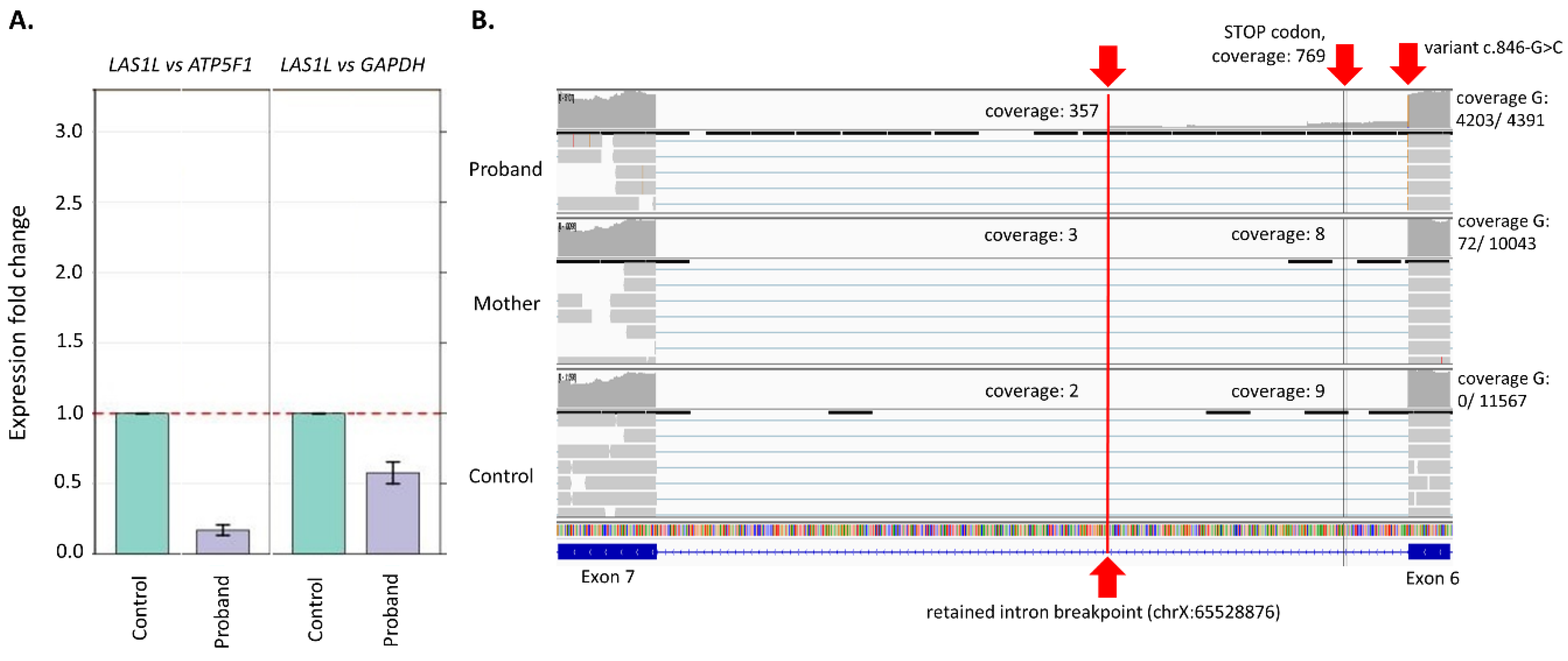
3.2. Gene Expression (qPCR)
4. Results
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| WES | Whole Exome Sequencing |
| OMIM | Online Mendelian Inheritance in Man |
| MRI | Magnetic resonance imaging |
| MS–MLPA | Methylation-Specific Multiplex Ligation-dependent Probe Amplification |
| PWS | Prader Willi Syndrome |
| AVAPS | Average volume-assured pressure support |
| ACMG | American College of Medical Genetics and Genomics |
| NMD | Nonsense-Mediated Decay |
References
- Hu, H.; Haas, S.A.; Chelly, J.; Van Esch, H.; Raynaud, M.; de Brouwer, A.P.; Weinert, S.; Froyen, G.; Frints, S.G.; Laumonnier, F.; et al. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol. Psychiatry 2016, 21, 133–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertini, E.; Houlden, H. Defects of RNA metabolism in the pathogenesis of spinal muscular atrophy. Neurology 2014, 82, 1298–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterfield, R.J.; Stevenson, T.J.; Xing, L.; Newcomb, T.M.; Nelson, B.; Zeng, W.; Li, X.; Lu, H.M.; Lu, H.; Gonzalez, K.D.F.; et al. Congenital lethal motor neuron disease with a novel defect in ribosome biogenesis. Neurology 2014, 82, 1322–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perego, M.G.L.; Galli, N.; Nizzardo, M.; Govoni, A.; Taiana, M.; Bresolin, N.; Comi, G.P.; Corti, S. Current understanding of and emerging treatment options for spinal muscular atrophy with respiratory distress type 1 (SMARD1). Cell. Mol. Life Sci. 2020, 77, 3351–3367. [Google Scholar] [CrossRef] [PubMed]
- Ramser, J.; Ahearn, M.E.; Lenski, C.; Yariz, K.O.; Hellebrand, H.; von Rhein, M.; Clark, R.D.; Schmutzler, R.K.; Lichtner, P.; Hoffman, E.P.; et al. Rare missense and synonymous variants in UBE1 are associated with X-linked infantile spinal muscular atrophy. Am. J. Hum. Genet. 2008, 82, 188–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saladini, M.; Nizzardo, M.; Govoni, A.; Taiana, M.; Bresolin, N.; Comi, G.P.; Corti, S. Spinal muscular atrophy with respiratory distress type 1: Clinical phenotypes, molecular pathogenesis and therapeutic insights. J. Cell. Mol. Med. 2020, 24, 1169–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rydzanicz, M.; Wachowska, M.; Cook, E.C.; Lisowski, P.; Kuźniewska, B.; Szymańska, K.; Diecke, S.; Prigione, A.; Szczałuba, K.; Szybińska, A.; et al. Novel calcineurin A (PPP3CA) variant associated with epilepsy, constitutive enzyme activation and downregulation of protein expression. Eur. J. Hum. Genet. 2019, 27, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Jian, X.; Boerwinkle, E.; Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014, 42, 13534–13544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San Millan, B.; Fernandez, J.M.; Navarro, C.; Reparaz, A.; Teijeira, S. Spinal muscular atrophy with respiratory distress type 1 (SMARD1) Report of a Spanish case with extended clinicopathological follow-up. Clin. Neuropathol. 2016, 35, 58–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schottmann, G.; Jungbluth, H.; Schara, U.; Knierim, E.; Morales Gonzalez, S.; Gill, E.; Seifert, F.; Norwood, F.; Deshpande, C.; von Au, K.; et al. Recessive truncating IGHMBP2 mutations presenting as axonal sensorimotor neuropathy. Neurology 2015, 84, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Pedurupillay, C.R.; Amundsen, S.S.; Barøy, T.; Rasmussen, M.; Blomhoff, A.; Stadheim, B.F.; Ørstavik, K.; Holmgren, A.; Iqbal, T.; Frengen, E.; et al. Clinical and molecular characteristics in three families with biallelic mutations in IGHMBP2. Neuromuscul. Disord. 2016, 26, 570–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grohmann, K.; Varon, R.; Stolz, P.; Schuelke, M.; Janetzki, C.; Bertini, E.; Bushby, K.; Muntoni, F.; Ouvrier, R.; Van Maldergem, L.; et al. Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Ann. Neurol. 2003, 54, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Jędrzejowska, M.; Madej-Pilarczyk, A.; Fidziańska, A.; Mierzewska, H.; Pronicka, E.; Obersztyn, E.; Gos, M.; Pronicki, M.; Kmieć, T.; Migdał, M.; et al. Severe phenotypes of SMARD1 associated with novel mutations of the IGHMBP2 gene and nuclear degeneration of muscle and Schwann cells. Eur. J. Paediatr. Neurol. 2014, 18, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Giannini, A.; Pinto, A.M.; Rossetti, G.; Prandi, E.; Tiziano, D.; Brahe, C.; Nardocci, N. Respiratory failure in infants due to spinal muscular atrophy with respiratory distress type 1. Intensive Care Med. 2006, 32, 1851–1855. [Google Scholar] [CrossRef] [PubMed]
- Guenther, U.P.; Varon, R.; Schlicke, M.; Dutrannoy, V.; Volk, A.; Hübner, C.; von Au, K.; Schuelke, M. Clinical and mutational profile in spinal muscular atrophy with respiratory distress (SMARD): Defining novel phenotypes through hierarchical cluster analysis. Hum. Mutat. 2007, 28, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Pitt, M.; Houlden, H.; Jacobs, J.; Mok, Q.; Harding, B.; Reilly, M.; Surtees, R. Severe infantile neuropathy with diaphragmatic weakness and its relationship to SMARD1. Brain 2003, 126 Pt 12, 2682–2692. [Google Scholar] [CrossRef]
- Eckart, M.; Guenther, U.P.; Idkowiak, J.; Varon, R.; Grolle, B.; Boffi, P.; Van Maldergem, L.; Hübner, C.; Schuelke, M.; von Au, K. The natural course of infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Pediatrics 2012, 129, e148–e156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viguier, A.; Lauwers-Cances, V.; Cintas, P.; Manel, V.; Peudenier, S.; Desguerre, I.; Quijano-Roy, S.; Vanhulle, C.; Fradin, M.; Isapof, A.; et al. Spinal muscular atrophy with respiratory distress type 1: A multicenter retrospective study. Neuromuscul. Disord. 2019, 29, 114–126. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stembalska, A.; Rydzanicz, M.; Walas, W.; Gasperowicz, P.; Pollak, A.; Pienkowski, V.M.; Biela, M.; Klaniewska, M.; Gamrot, Z.; Gronska, E.; et al. Severe Infantile Axonal Neuropathy with Respiratory Failure Caused by Novel Mutation in X-Linked LAS1L Gene. Genes 2022, 13, 725. https://doi.org/10.3390/genes13050725
Stembalska A, Rydzanicz M, Walas W, Gasperowicz P, Pollak A, Pienkowski VM, Biela M, Klaniewska M, Gamrot Z, Gronska E, et al. Severe Infantile Axonal Neuropathy with Respiratory Failure Caused by Novel Mutation in X-Linked LAS1L Gene. Genes. 2022; 13(5):725. https://doi.org/10.3390/genes13050725
Chicago/Turabian StyleStembalska, Agnieszka, Małgorzata Rydzanicz, Wojciech Walas, Piotr Gasperowicz, Agnieszka Pollak, Victor Murcia Pienkowski, Mateusz Biela, Magdalena Klaniewska, Zuzanna Gamrot, Ewa Gronska, and et al. 2022. "Severe Infantile Axonal Neuropathy with Respiratory Failure Caused by Novel Mutation in X-Linked LAS1L Gene" Genes 13, no. 5: 725. https://doi.org/10.3390/genes13050725
APA StyleStembalska, A., Rydzanicz, M., Walas, W., Gasperowicz, P., Pollak, A., Pienkowski, V. M., Biela, M., Klaniewska, M., Gamrot, Z., Gronska, E., Ploski, R., & Smigiel, R. (2022). Severe Infantile Axonal Neuropathy with Respiratory Failure Caused by Novel Mutation in X-Linked LAS1L Gene. Genes, 13(5), 725. https://doi.org/10.3390/genes13050725