
Genomic and Transcriptomic Characterization of Atypical Recurrent Flank Alopecia in the Cesky Fousek

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Blood Sample Collection
2.2. Biopsy Sample Collection
2.3. Genotyping
2.4. Case/Control GWAS
2.5. Quantitative GWAS (QGWAS) and Additional GWAS Analyses
2.6. Haplotype Identification
2.7. Histopathological Analysis
2.8. RNA Extraction and RNA-Seq Experiments
2.9. Differential Expression Analysis
2.10. Protein–Protein Interaction Analysis
3. Results
3.1. Population Genetic Structure of aRFA Affected and Control Individuals
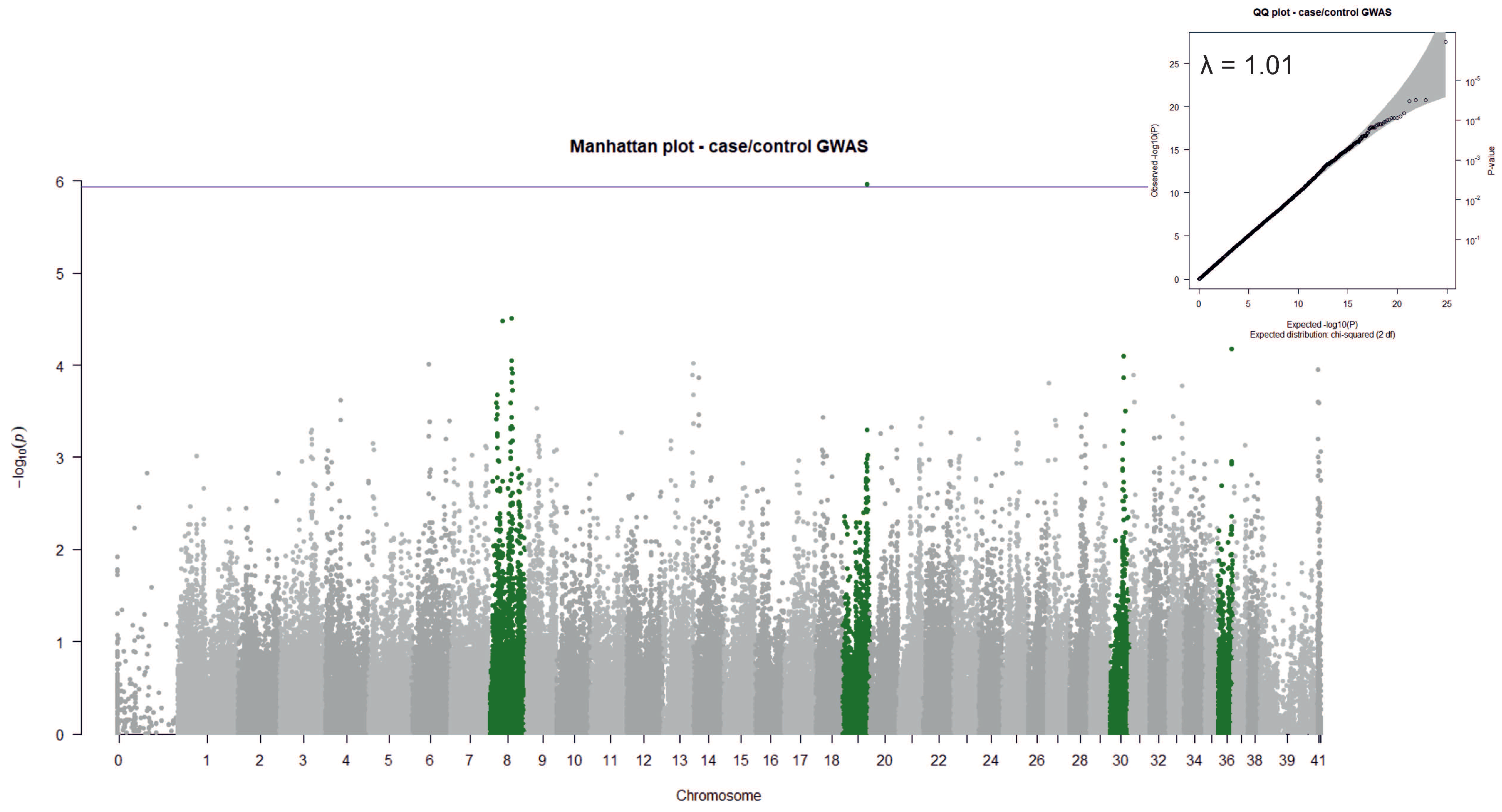
3.2. Case/Control GWAS
3.3. Quantitative GWAS and Additional GWAS Analyses
3.4. Haplotype Identification
3.5. Candidate Genes Identified by GWAS
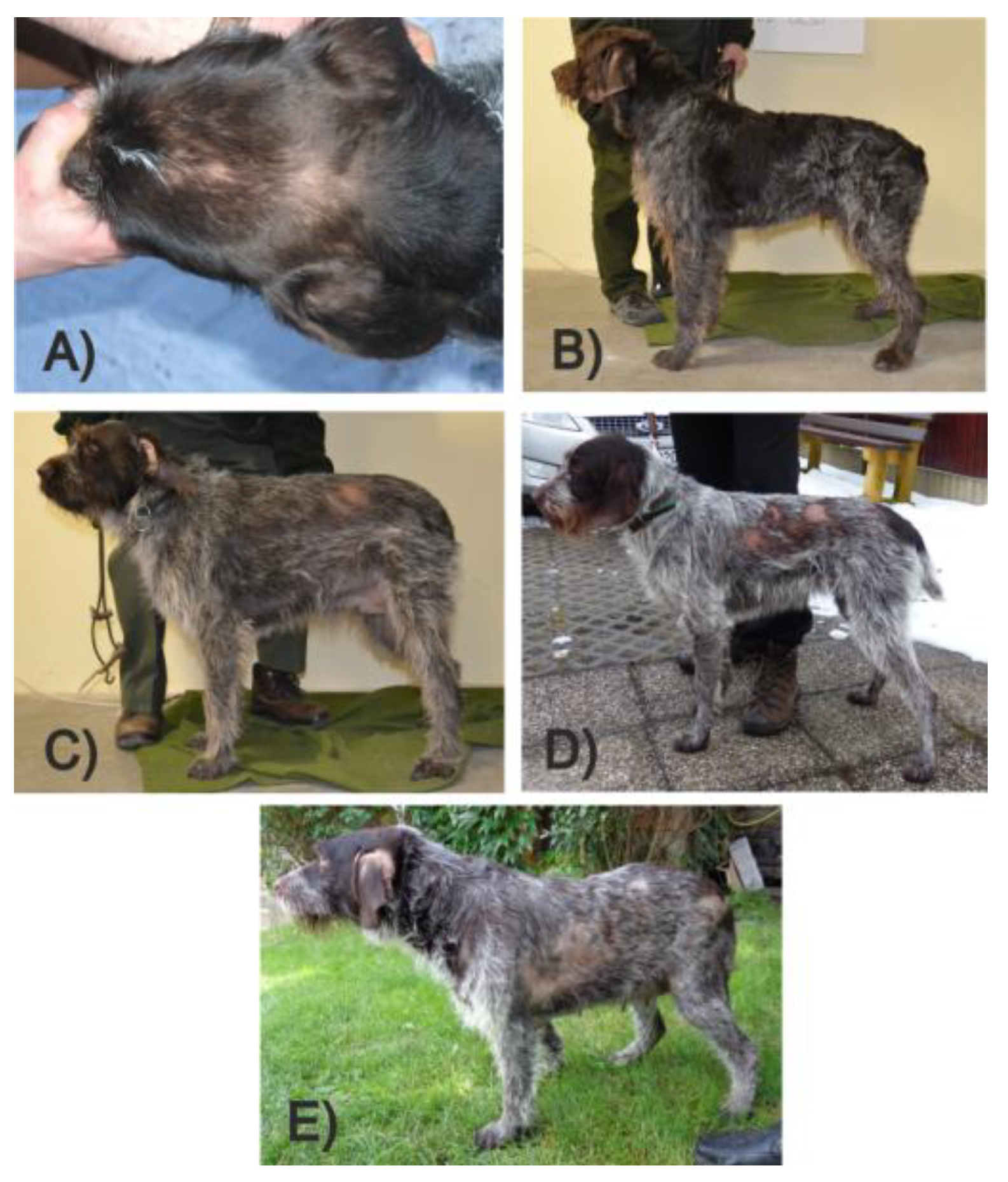
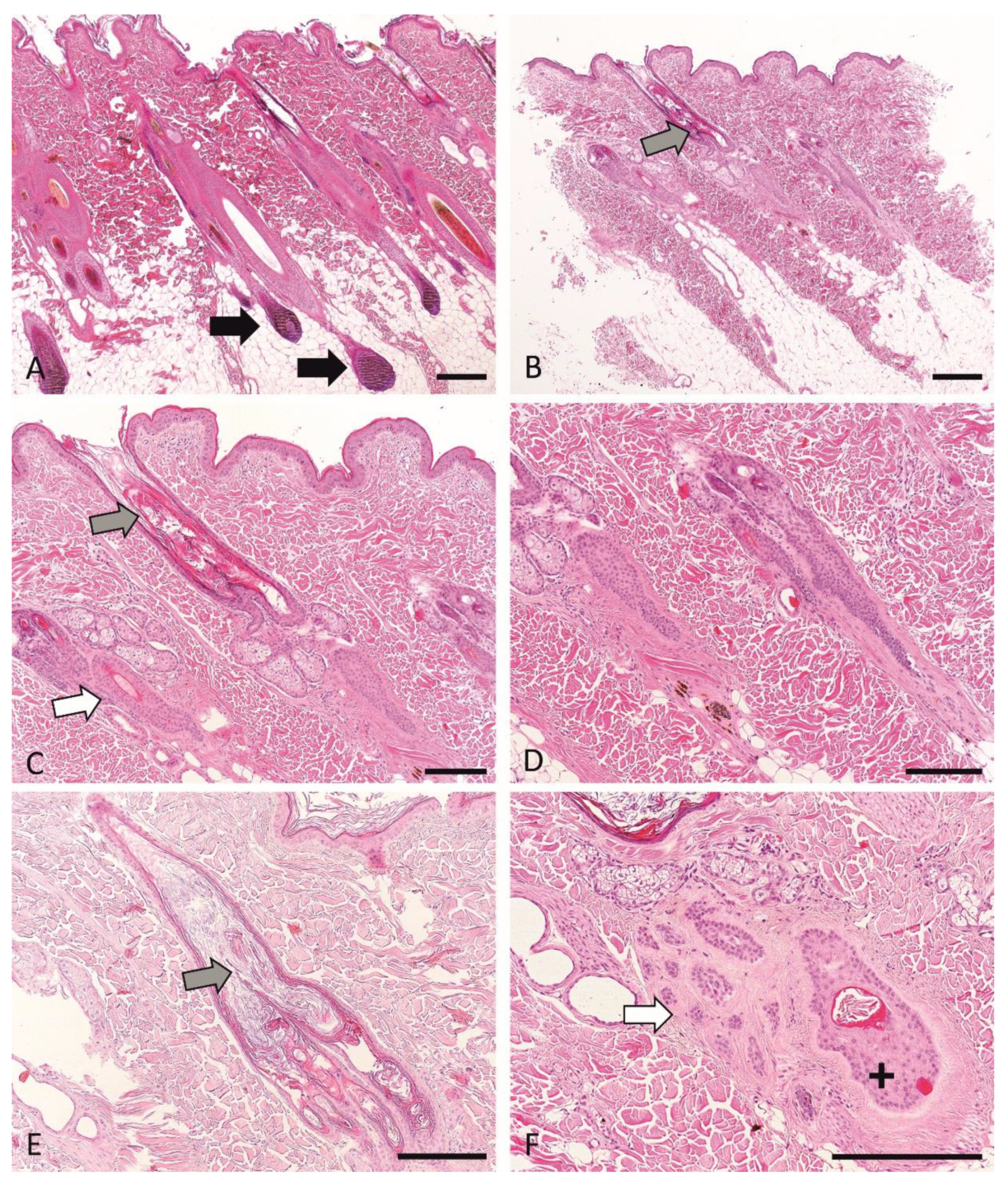
3.6. Histopathological Phenotyping and Sample Selection
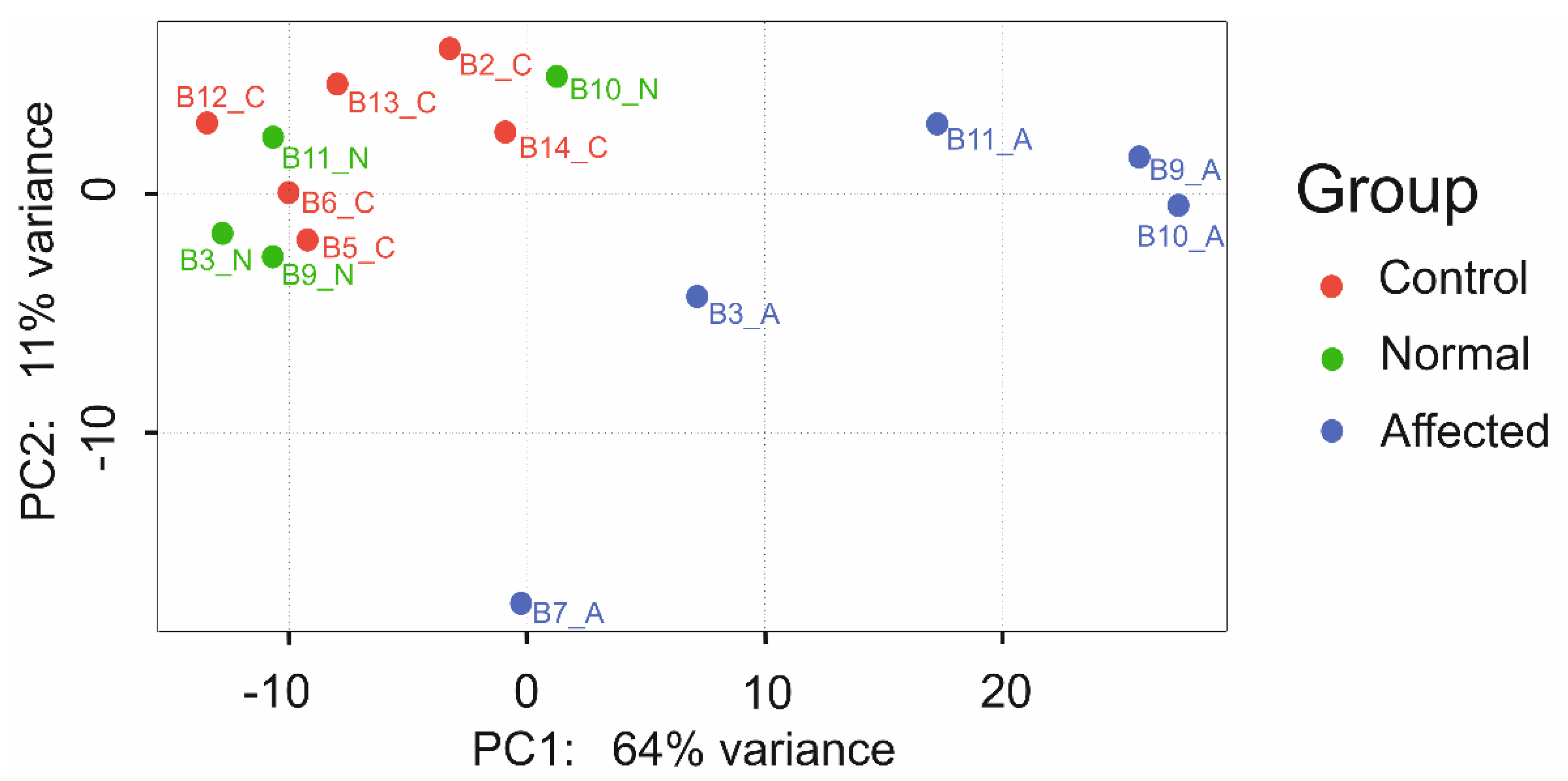
3.7. RNA Sequencing Analysis
3.8. Functional Classification of DEGs
3.9. Protein–protein Interaction Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shearin, A.L.; Ostrander, E.A. Leading the way: Canine models of genomics and disease. Dis. Models Mech. 2010, 3, 27–34. [Google Scholar] [CrossRef]
- Hayward, J.J.; Castelhano, M.G.; Oliveira, K.C.; Corey, E.; Balkman, C.; Baxter, T.L.; Casal, M.L.; Center, S.A.; Fang, M.; Garrison, S.J.; et al. Complex disease and phenotype mapping in the domestic dog. Nat. Commun. 2016, 7, 10460. [Google Scholar] [CrossRef] [PubMed]
- Steenbeek, F.G.; van Hytönen, M.K.; Leegwater, P.A.J.; Lohi, H. The canine era: The rise of a biomedical model. Anim. Genet. 2016, 47, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Lequarré, A.-S.; Andersson, L.; André, C.; Fredholm, M.; Hitte, C.; Leeb, T.; Lohi, H.; Lindblad-Toh, K.; Georges, M. LUPA: A European initiative taking advantage of the canine genome architecture for unravelling complex disorders in both human and dogs. Vet. J. 2011, 189, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Marsden, C.D.; Ortega-Del Vecchyo, D.; O’Brien, D.P.; Taylor, J.F.; Ramirez, O.; Vilà, C.; Marques-Bonet, T.; Schnabel, R.D.; Wayne, R.K.; Lohmueller, K.E. Bottlenecks and selective sweeps during domestication have increased deleterious genetic variation in dogs. Proc. Natl. Acad. Sci. USA 2016, 113, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, E.K.; Baranowska, I.; Wade, C.M.; Salmon Hillbertz, N.H.C.; Zody, M.C.; Anderson, N.; Biagi, T.M.; Patterson, N.; Pielberg, G.R.; Kulbokas, E.J.; et al. Efficient mapping of mendelian traits in dogs through genome-wide association. Nat. Genet. 2007, 39, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Chuong, C.M. Multi-layered environmental regulation on the homeostasis of stem cells: The saga of hair growth and alopecia. J. Dermatol. Sci. 2012, 66, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.R.; Schmidt-Ullrich, R.; Paus, R. The hair follicle as a dynamic miniorgan. Curr. Biol. 2009, 19, R132–R142. [Google Scholar] [CrossRef]
- Fuchs, E. Scratching the surface of skin development. Nature 2007, 445, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Ullrich, R.; Paus, R. Molecular principles of hair follicle induction and morphogenesis. Bioessays 2005, 27, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Wang, L.; Plikus, M.V.; Jiang, T.X.; Murray, P.J.; Ramos, R.; Guerrero-Juarez, C.F.; Hughes, M.W.; Lee, O.K.; Shi, S.; et al. Organ-level quorum sensing directs regeneration in hair stem cell populations. Cell 2015, 161, 277–290. [Google Scholar] [CrossRef]
- Hsu, Y.-C.; Pasolli, H.A.; Fuchs, E. Dynamics between stem cells, niche, and progeny in the hair follicle. Cell 2011, 144, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.V.; Cheong, J.; Ciapurin, N.; McDermitt, D.J.; Tumbar, T. Distinct self-renewal and differentiation phases in the niche of infrequently dividing hair follicle stem cells. Cell Stem Cell 2009, 5, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Rompolas, P.; Greco, V. Stem cell dynamics in the hair follicle niche. Semin. Cell Dev. Biol. 2014, 25–26, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Mesler, A.L.; Veniaminova, N.A.; Lull, M.V.; Wong, S.Y. Hair follicle terminal differentiation is orchestrated by distinct early and late matrix progenitors. Cell Rep. 2017, 19, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Milner, Y.; Kashgarian, M.; Sudnik, J.; Filippi, M.; Kizoulis, M.; Stenn, K. Exogen, shedding phase of the hair growth cycle: Characterization of a mouse model. J. Investig. Dermatol. 2002, 119, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Street, S.L.; Gaide, O.; Hertig, S.; Tardivel, A.; Tschopp, J.; Runkel, L.; Alevizopoulos, K.; Ferguson, B.M.; Zonana, J. Mutations leading to x-linked hypohidrotic ectodermal dysplasia affect three major functional domains in the tumor necrosis factor family member ectodysplasin-a. J. Biol. Chem. 2001, 276, 18819–18827. [Google Scholar] [CrossRef] [PubMed]
- Casal, M.L.; Jezyk, P.F.; Greek, J.M.; Goldschmidt, M.H.; Patterson, D.F. X-linked ectodermal dysplasia in the dog. J. Hered. 1997, 88, 513–517. [Google Scholar] [CrossRef][Green Version]
- Casal, M.L.; Scheidt, J.L.; Rhodes, J.L.; Henthorn, P.S.; Werner, P. Mutation Identification in a canine model of x-linked ectodermal dysplasia. Mamm. Genome 2005, 16, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Waluk, D.P.; Zur, G.; Kaufmann, R.; Welle, M.M.; Jagannathan, V.; Drögemüller, C.; Müller, E.J.; Leeb, T.; Galichet, A. A splice defect in the eda gene in dogs with an x-linked hypohidrotic ectodermal dysplasia (XLHED) phenotype. G3 Genes Genomes Genet. 2016, 6, 2949–2954. [Google Scholar] [CrossRef]
- Drögemüller, C.; Karlsson, E.K.; Hytönen, M.K.; Perloski, M.; Dolf, G.; Sainio, K.; Lohi, H.; Lindblad-Toh, K.; Leeb, T. A mutation in hairless dogs implicates Foxi3 in ectodermal development. Science 2008, 321, 1462. [Google Scholar] [CrossRef]
- Frank, L.A.; Hnilica, K.A.; Oliver, J.W. Adrenal steroid hormone concentrations in dogs with hair cycle arrest (Alopecia X) before and during treatment with melatonin and mitotane. Vet. Dermatol. 2004, 15, 278–284. [Google Scholar] [CrossRef]
- Cerundolo, R. Symmetrical alopecia in the dog. Practice 1999, 21, 350–359. [Google Scholar] [CrossRef]
- Mausberg, E.-M.; Drögemüller, C.; Dolf, G.; Leeb, T.; Rüfenacht, S.; Welle, M. Evaluation of the CTSL2 gene as a candidate gene for alopecia x in pomeranians and keeshonden. Anim. Biotechnol. 2007, 18, 291–296. [Google Scholar] [CrossRef]
- Brunner, M.A.T.; Jagannathan, V.; Waluk, D.P.; Roosje, P.; Linek, M.; Panakova, L.; Leeb, T.; Wiener, D.J.; Welle, M.M. Novel insights into the pathways regulating the canine hair cycle and their deregulation in alopecia x. PLoS ONE 2017, 12, e0186469. [Google Scholar] [CrossRef]
- Frank, L.A. Oestrogen receptor antagonist and hair regrowth in dogs with hair cycle arrest (Alopecia X). Vet. Dermatol. 2007, 18, 63–66. [Google Scholar] [CrossRef]
- Vandenabeele, S.; Declercq, J.; De Cock, H.; Daminet, S. Canine recurrent flank alopecia: A synthesis of theory and practice. Vlaams Diergeneeskd. Tijdschr. 2014, 83, 275–283. [Google Scholar] [CrossRef]
- Miller, M.A.; Dunstan, R.W. Seasonal flank alopecia in boxers and airedale terriers: 24 cases (1985–1992). J. Am. Vet. Med. Assoc. 1993, 203, 1567–1572. [Google Scholar]
- Bassett, R.; Burton, G.; Robson, D. Recurrent flank alopecia in a Tibetan terrier. Aust. Vet. J. 2005, 83, 276–279. [Google Scholar] [CrossRef]
- Waldman, L. Seasonal flank alopecia in Affenpinschers. J. Small Anim. Pract. 1995, 36, 271–273. [Google Scholar] [CrossRef]
- Gomes, L.A.; Sonoda, M.C.; Bortolucci, L.; Werner, J.; Headley, S.A. Manifestations of cyclical flank alopecia in a dog from Southern Brazil. Braz. J. Vet. Pathol. 2008, 5, 59–63. [Google Scholar]
- Paradis, M.; Cerundolo, R. An Approach to Symmetrical Alopecia in the Dog. In BSAVA Manual of Canine and Feline Dermatology; BSAVA Library: Gloucester, UK, 2012; ISBN 978-1-905319-88-6. [Google Scholar]
- Medleau, L.; Hnilica, K.A. Small Animal Dermatology: A Color Atlas and Therapeutic Guide, 2nd ed.; Saunders: Philadelphia, PA, USA, 2006. [Google Scholar]
- Curtis, C.F.; Evans, H.; Lloyd, D.H. Investigation of the Reproductive and growth hormone status of dogs affected by idiopathic recurrent flank alopecia. J. Small Anim. Pract. 1996, 37, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Havlát, J. Seznam Chovných Psů Pro Rok 2022/List of Stud Males for 2022. Cesky Fousek Breeding Club Home Page. 2022. Available online: https://ceskyfousekna.org/ (accessed on 19 February 2022).
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Purcell, S. PLINK 1.07. Available online: http://pngu.mgh.harvard.edu/purcell/plink/ (accessed on 30 January 2020).
- Hayward, J.J.; Kelly-Smith, M.; Boyko, A.R.; Burmeister, L.; De Risio, L.; Mellersh, C.; Freeman, J.; Strain, G.M. A genome-wide association study of deafness in three canine breeds. PLoS ONE 2020, 15, e0232900. [Google Scholar] [CrossRef]
- Tsai, K.L.; Evans, J.M.; Noorai, R.E.; Starr-Moss, A.N.; Clark, L.A. Novel Y Chromosome retrocopies in canids revealed through a genome-wide association study for sex. Genes 2019, 10, 320. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal Components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Patterson, N.; Price, A.L.; Reich, D. Population structure and eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.R-Project.Org/ (accessed on 19 February 2022).
- Clayton, D. snpStats: SnpMatrix and XSnpMatrix Classes and Methods. 2021. Available online: https://bioconductor.org/packages/release/bioc/html/snpStats.html (accessed on 19 February 2022). [CrossRef]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Kluyver, T.; Ragan-Kelley, B.; Pérez, F.; Granger, B.; Bussonnier, M.; Frederic, J. Jupyter Notebooks–A Publishing Format for Reproducible Computational Workflows. In Positioning and Power in Academic Publishing: Players, Agents and Agendas; IOS Press: Amsterdam, The Netherlands, 2016; pp. 87–90. [Google Scholar]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef]
- Purcell, S.; Chang, C. PLINK 1.9. Available online: https://www.cog-genomics.org/plink/1.9/ (accessed on 30 January 2020).
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Delaneau, O.; Howie, B.; Cox, A.J.; Zagury, J.-F.; Marchini, J. Haplotype estimation using sequencing reads. Am. J. Hum. Genet. 2013, 93, 687–696. [Google Scholar] [CrossRef]
- Brunner, M.A.T.; Rüfenacht, S.; Bauer, A.; Erpel, S.; Buchs, N.; Braga-Lagache, S.; Heller, M.; Leeb, T.; Jagannathan, V.; Wiener, D.J.; et al. Bald thigh syndrome in sighthounds—Revisiting the cause of a well-known disease. PLoS ONE 2019, 14, e0212645. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING V10: Protein–Protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Bohutínská, M.; Alston, M.; Monnahan, P.; Mandáková, T.; Bray, S.; Paajanen, P.; Kolář, F.; Yant, L. Novelty and convergence in adaptation to whole genome duplication. Mol. Biol. Evol. 2021, 38, 3910–3924. [Google Scholar] [CrossRef] [PubMed]
- Neradilová, S.; Connell, L.; Hulva, P.; Černá Bolfíková, B. Tracing genetic resurrection of pointing dog breeds: Cesky Fousek as both survivor and rescuer. PLoS ONE 2019, 14, e0221418. [Google Scholar] [CrossRef]
- Wiener, D.J.; Groch, K.R.; Brunner, M.A.T.; Leeb, T.; Jagannathan, V.; Welle, M.M. Transcriptome Profiling and differential gene expression in canine microdissected anagen and telogen hair follicles and interfollicular epidermis. Genes 2020, 11, 884. [Google Scholar] [CrossRef] [PubMed]
- Dostál, J.; Martenek, M.; Tripes, O.; Koberová, S. Český Fousek; Chováme Psy; Dona: České Budějovice, Czech Republic, 1998; ISBN 978-80-85463-97-2. [Google Scholar]
- Sheet, S.; Kim, J.-S.; Ko, M.-J.; Kim, N.-Y.; Lim, Y.-J.; Park, M.-R.; Lee, S.-J.; Kim, J.-M.; Oh, S.-I.; Choi, B.-H. Insight into the candidate genes and enriched pathways associated with height, length, length to height ratio and body-weight of Korean indigenous breed, Jindo dog using gene set enrichment-based GWAS analysis. Animals 2021, 11, 3136. [Google Scholar] [CrossRef]
- Hagenaars, S.P.; Hill, W.D.; Harris, S.E.; Ritchie, S.J.; Davies, G.; Liewald, D.C.; Gale, C.R.; Porteous, D.J.; Deary, I.J.; Marioni, R.E. Genetic prediction of male pattern baldness. PLoS Genet. 2017, 13, e1006594. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Noh, H.J.; Wang, D.; Sigurdsson, S.; Swofford, R.; Perloski, M.; Duxbury, M.; Patterson, E.E.; Albright, J.; Castelhano, M.; et al. Candidate genes and functional noncoding variants identified in a canine model of obsessive-compulsive disorder. Genome Biol. 2014, 15, R25. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Stenn, K.S.; Paus, R. Controls of Hair Follicle Cycling. Physiol. Rev. 2001, 81, 449–494. [Google Scholar] [CrossRef]
- Almohanna, H.M.; Ahmed, A.A.; Tsatalis, J.P.; Tosti, A. The role of vitamins and minerals in hair loss: A review. Dermatol. Ther. 2019, 9, 51–70. [Google Scholar] [CrossRef]
- Lin, K.K.; Kumar, V.; Geyfman, M.; Chudova, D.; Ihler, A.T.; Smyth, P.; Paus, R.; Takahashi, J.S.; Andersen, B. Circadian clock genes contribute to the regulation of hair follicle cycling. PLoS Genet. 2009, 5, e1000573. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Plikus, M.V.; Tang, P.-C.; Widelitz, R.B.; Chuong, C.M. The modulatable stem cell niche: Tissue interactions during hair and feather follicle regeneration. J. Mol. Biol. 2016, 428, 1423–1440. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Tumbar, T. Hairy tale of signaling in hair follicle development and cycling. Semin. Cell Dev. Biol. 2012, 23, 906–916. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, K.; Clevers, H. Wnt/β-catenin signaling in adult mammalian epithelial stem cells. Dev. Biol. 2017, 428, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Tanaka, N. Roles of the hedgehog signaling pathway in epidermal and hair follicle development, homeostasis, and cancer. J. Dev. Biol. 2017, 5, 12. [Google Scholar] [CrossRef]
- Kawano, Y.; Kypta, R. Secreted antagonists of the Wnt signalling pathway. J. Cell Sci. 2003, 116, 2627–2634. [Google Scholar] [CrossRef] [PubMed]
- Greco, V.; Chen, T.; Rendl, M.; Schober, M.; Pasolli, H.A.; Stokes, N.; dela Cruz-Racelis, J.; Fuchs, E. A two-step mechanism for stem cell activation during hair regeneration. Cell Stem Cell 2009, 4, 155–169. [Google Scholar] [CrossRef]
- Hsu, Y.-C.; Li, L.; Fuchs, E. Transit-amplifying cells orchestrate stem cell activity and tissue regeneration. Cell 2014, 157, 935–949. [Google Scholar] [CrossRef]
- Welle, M.M.; Wiener, D.J. The hair follicle: A comparative review of canine hair follicle anatomy and physiology. Toxicol. Pathol. 2016, 44, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Stenn, K.S.; Karnik, P. Lipids to the top of hair biology. J. Investig. Dermatol. 2010, 130, 1205–1207. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R. Wnts and hedgehogs: Lipid-modified proteins and similarities in signaling mechanisms at the cell surface. Development 2003, 130, 5297–5305. [Google Scholar] [CrossRef] [PubMed]
- Evers, A.W.M.; Verhoeven, E.W.M.; Kraaimaat, F.W.; Jong, E.M.G.J.D.; Brouwer, S.J.M.D.; Schalkwijk, J.; Sweep, F.C.G.J.; Kerkhof, P.C.M.V.D. How stress gets under the skin: Cortisol and stress reactivity in psoriasis. Br. J. Dermatol. 2010, 163, 986–991. [Google Scholar] [CrossRef]
- Morinaga, H.; Mohri, Y.; Grachtchouk, M.; Asakawa, K.; Matsumura, H.; Oshima, M.; Takayama, N.; Kato, T.; Nishimori, Y.; Sorimachi, Y.; et al. Obesity accelerates hair thinning by stem cell-centric converging mechanisms. Nature 2021, 595, 266–271. [Google Scholar] [CrossRef]
- Dall’Aglio, C.; Maranesi, M.; Di Loria, A.; Piantedosi, D.; Ciaramella, P.; Alterisio, M.C.; Lepri, E.; Mercati, F. Effects of obesity on adiponectin system skin expression in dogs: A comparative study. Animals 2021, 11, 2308. [Google Scholar] [CrossRef]
- Palmer, M.A.; Blakeborough, L.; Harries, M.; Haslam, I.S. Cholesterol homeostasis: Links to hair follicle biology and hair disorders. Exp. Dermatol. 2020, 29, 299–311. [Google Scholar] [CrossRef]
- Spustová, V.; Dzúrik, R. Vitamin D: Synthesis, metabolism, regulation, and an assessment of its deficiency in patients with chronic renal disease. Vnitr. Lek. 2004, 50, 537–543. [Google Scholar]
- Sakai, Y.; Kishimoto, J.; Demay, M.B. Metabolic and cellular analysis of alopecia in vitamin D receptor knockout mice. J. Clin. Investig. 2001, 107, 961–966. [Google Scholar] [CrossRef]
- Bikle, D.D. Vitamin D and the skin: Physiology and pathophysiology. Rev. Endocr. Metab. Disord. 2012, 13, 3–19. [Google Scholar] [CrossRef]
- Demay, M.B.; MacDonald, P.N.; Skorija, K.; Dowd, D.R.; Cianferotti, L.; Cox, M. Role of the vitamin D receptor in hair follicle biology. J. Steroid Biochem. Mol. Biol. 2007, 103, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-H.; Lee, J.-E.; An, S.-M.; Shin, Y.Y.; Hwang, D.Y.; Yang, S.Y.; Cho, S.-K.; An, B.-S. Effect of vitamin D3 on biosynthesis of estrogen in porcine granulosa cells via modulation of steroidogenic enzymes. Toxicol. Res. 2017, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, R. Pathogenesis and Treatment of Alopecia in Humans. In Proceedings of the World Congress of Veterinary Dermatology, Sydney, Australia, 20–24 October 2020; Volume 9, p. 13. [Google Scholar]
- Ge, Y.; Miao, Y.; Gur-Cohen, S.; Gomez, N.; Yang, H.; Nikolova, M.; Polak, L.; Hu, Y.; Verma, A.; Elemento, O.; et al. The aging skin microenvironment dictates stem cell behavior. Proc. Natl. Acad. Sci. USA 2020, 117, 5339–5350. [Google Scholar] [CrossRef]
- Lee, S.A.; Li, K.N.; Tumbar, T. Stem cell-intrinsic mechanisms regulating adult hair follicle homeostasis. Exp. Dermatol. 2021, 30, 430–447. [Google Scholar] [CrossRef] [PubMed]
- Mokry, J.; Pisal, R. Development and maintenance of epidermal stem cells in skin adnexa. Int. J. Mol. Sci. 2020, 21, 9736. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr | SNP Name | Position (bp) | Allele Freq | p-Value |
|---|---|---|---|---|
| 19 | BICF2G630255452 | 47,856,573 | 0.333 | 1.08 × 10−6 |
| 8 | BICF2P465820 | 43,487,284 | 0.262 | 3.10 × 10−5 |
| 8 | TIGRP2P114211_rs8542415 | 434,942,31 | 0.262 | 3.10 × 10−5 |
| 8 | BICF2S23110497 | 25,810,719 | 0.205 | 3.30 × 10−5 |
| 36 | BICF2P1194573 | 28,584,717 | 0.271 | 6.72 × 10−5 |
| 30 | BICF2G630401492 | 26,273,661 | 0.326 | 8.07 × 10−5 |
| 8 | BICF2P361090 | 43,341,287 | 0.233 | 8.90 × 10−5 |
| 8 | BICF2P543725 | 43,371,261 | 0.233 | 8.90 × 10−5 |
| 8 | BICF2S23137831 | 43,418,611 | 0.233 | 8.89 × 10−5 |
| 13 | BICF2P281837 | 63,012,417 | 0.057 | 9.61 × 10−5 |
| 6 | BICF2P742566 | 35,078,147 | 0.309 | 9.86 × 10−5 |
| 8 | BICF2P177234 | 43,520,222 | 0.235 | 1.10 × 10−4 |
| 41 | BICF2S23546044 | 18,45,101 | 0.310 | 1.11 × 10−4 |
| 8 | TIGRP2P114933_rs9187625 | 46,799,348 | 0.493 | 1.24 × 10−4 |
| 31 | BICF2P1368177 | 7,605,782 | 0.104 | 1.27 × 10−4 |
| 31 | BICF2S2443709 | 7,615,165 | 0.104 | 1.27 × 10−4 |
| 13 | BICF2G630745860 | 61,855,230 | 0.149 | 1.28 × 10−4 |
| 14 | BICF2G630521203 | 10,825,554 | 0.061 | 1.36 × 10−4 |
| 30 | TIGRP2P370921_rs8763952 | 26,977,673 | 0.233 | 1.36 × 10−4 |
| 8 | BICF2P1102123 | 43,411,814 | 0.255 | 1.54 × 10−4 |
| Chr (SNP) | Genotype | No. Controls | % Controls | No. Cases | % Cases |
|---|---|---|---|---|---|
| chr19 (BICF2G630255452) | AA | 69 | 59.0 | 26 | 26.8 |
| GA | 41 | 35.0 | 53 | 54.6 | |
| GG | 7 | 6.0 | 17 | 18.6 | |
| chr8 (BICF2P465820) | AA | 16 | 13.7 | 1 | 1.0 |
| AG | 50 | 42.7 | 27 | 27.8 | |
| GG | 51 | 43.6 | 68 | 70.1 |
| Chr | SNP Name | Position (bp) | Allele Freq | p-Value |
|---|---|---|---|---|
| 8 | BICF2P361090 | 43,341,287 | 0.235 | 4.81 × 10−6 |
| 8 | BICF2P543725 | 43,371,261 | 0.235 | 4.81 × 10−6 |
| 8 | BICF2S23137831 | 43,418,611 | 0.235 | 4.81 × 10−6 |
| 8 | BICF2P465820 | 43,487,284 | 0.263 | 5.56 × 10−6 |
| 8 | TIGRP2P114211_rs8542415 | 43,494,231 | 0.263 | 5.56 × 10−6 |
| 8 | BICF2S23110497 | 25,810,719 | 0.207 | 5.81 × 10−6 |
| 8 | BICF2P177234 | 43,520,222 | 0.236 | 7.36 × 10−6 |
| 19 | BICF2G630255452 | 47,856,573 | 0.335 | 9.05 × 10−6 |
| 8 | TIGRP2P114933_rs9187625 | 46,799,348 | 0.495 | 1.36 × 10−5 |
| 8 | BICF2S23235533 | 15,314,523 | 0.251 | 1.53 × 10−5 |
| 8 | BICF2S22921051 | 15,005,970 | 0.260 | 1.56 × 10−5 |
| 8 | BICF2P1102123 | 43,411,814 | 0.256 | 1.85 × 10−5 |
| 8 | BICF2P1109401 | 43,462,069 | 0.256 | 1.85 × 10−5 |
| 8 | BICF2P146090 | 43,425,554 | 0.256 | 1.85 × 10−5 |
| 8 | BICF2P396875 | 43,463,543 | 0.256 | 1.85 × 10−5 |
| 8 | BICF2P755461 | 43,441,286 | 0.256 | 1.85 × 10−5 |
| 8 | BICF2P762487 | 43,454,904 | 0.256 | 1.85 × 10−5 |
| 8 | BICF2S22932019 | 46,809,268 | 0.493 | 2.18 × 10−5 |
| 31 | BICF2P1368177 | 7,605,782 | 0.102 | 2.58 × 10−5 |
| 31 | BICF2S2443709 | 7,615,165 | 0.102 | 2.58 × 10−5 |
| Chr (SNP) | Genotype | Healthy (%) | Head (%) | L1 (%) | L2 (%) | L3 (%) | L4 (%) |
|---|---|---|---|---|---|---|---|
| chr8 (BICF2P361090) | AA | 52 (47) | 1 (33) | 4 (67) | 14 (50) | 43 (88) | 15 (79) |
| CA | 46 (41) | 2 (67) | 1 (17) | 13 (46) | 5 (10) | 4 (21) | |
| CC | 13 (12) | 0 | 1 (17) | 0 | 1 (2) | 0 | |
| missing | 0 | 0 | 0 | 1 (4) | 0 | 0 | |
| total | 111 | 3 | 6 | 28 | 49 | 19 |
| Chr | SNP Name | Position (bp) | Allele Freq | p-Value |
|---|---|---|---|---|
| 21 | BICF2G630640798 | 47,085,771 | 0.221 | 5.01 × 10−7 |
| 23 | BICF2S23432401 | 11,113,618 | 0.377 | 5.75 × 10−6 |
| 37 | TIGRP2P420015_rs8709645 | 20,114,103 | 0.262 | 6.48 × 10−6 |
| 8 | chr8_59707832 | 59,707,832 | 0.221 | 8.66 × 10−6 |
| 37 | BICF2G630131116 | 25,669,986 | 0.148 | 1.07 × 10−5 |
| 15 | BICF2G630419811 | 59,659,531 | 0.434 | 1.22 × 10−5 |
| 23 | BICF2P438054 | 11,110,146 | 0.352 | 1.30 × 10−5 |
| 21 | BICF2G630641744 | 46,513,869 | 0.254 | 1.60 × 10−5 |
| 27 | BICF2G630139626 | 42,94,734 | 0.205 | 1.92 × 10−5 |
| 17 | chr17_40427743 | 40,427,743 | 0.426 | 2.87 × 10−5 |
| 21 | BICF2S23427379 | 46,584,445 | 0.270 | 3.07 × 10−5 |
| 20 | BICF2P1328442 | 55,962,058 | 0.320 | 3.49 × 10−5 |
| 27 | BICF2P675588 | 34,378,821 | 0.295 | 3.70 × 10−5 |
| 23 | BICF2G630386401 | 13,368,971 | 0.459 | 4.20 × 10−5 |
| 18 | BICF2G630699395 | 34,387,737 | 0.484 | 4.30 × 10−5 |
| 27 | BICF2G630139599 | 4,253,386 | 0.180 | 4.40 × 10−5 |
| 27 | BICF2G630139609 | 4,266,185 | 0.180 | 4.40 × 10−5 |
| 27 | BICF2G630139630 | 4,299,688 | 0.180 | 4.40 × 10−5 |
| 27 | BICF2G630139642 | 4,318,805 | 0.180 | 4.40 × 10−5 |
| 27 | BICF2S23028384 | 4,247,215 | 0.180 | 4.40 × 10−5 |
| Chr | bp | Haplotype | % Cases | % Controls | p-Value |
|---|---|---|---|---|---|
| 8 | 43,341,287–43,356,221 | AAG | 75.0 | 49.4 | 7.31 × 10−8 |
| 8 * | 43,463,820–43,494,231 | GGG | 85.0 | 62.4 | 2.25 × 10−7 |
| 19 | 19,807,697–20,172,164 | ATGGTCAGGG | 84.4 | 53.9 | 2.09 × 10−11 |
| 19 * | 47,856,573 | A | 54.7 | 76.5 | 2.03 × 10−6 |
| 19 * | 47,856,573 | G | 45.3 | 23.5 | 2.03 × 10−6 |
| 30 | 26,126,946–26,143,675 | GCGA | 15.8 | 35.5 | 5.04 × 10−6 |
| 30 * | 26,245,545–26,328,881 | ATACAGGA | 21.5 | 41.3 | 1.45 × 10−5 |
| 36 * | 28,573,704–28,584,717 | CC | 36.7 | 18.8 | 3.53 × 10−5 |
| Gene Symbol | Full Gene Name | Described Function | Signaling Pathway | BaseMean | Log2FC |
|---|---|---|---|---|---|
| CTNNB1 | catenin beta | promotes HF growth | WNT | 20,928.210 | −0.498 |
| CUX1 | Cutl1, cut like homeobox 1 | inhibitor of HF differentiation | NOTCH | 1600.827 | −0.895 |
| DLX1 | distal-less homeobox 1 | HF cycling and differentiation | WNT | 48.951 | −2.120 |
| DLX2 | distal-less homeobox 2 | HF cycling and differentiation | TGF-b | 32.891 | −1.852 |
| DLX3 | distal-less homeobox 3 | HF cycling and differentiation | WNT | 1315.187 | −1.154 |
| DLX5 | distal-less homeobox 5 | HF cycling and differentiation | BMP | 85.608 | 1.026 |
| FGF5 | fibroblast growth factor 5 | catagen induction | FGF | 124.674 | −2.896 |
| FOXE1 | forkhead box E1 | governs HF stem cell (SC) niche | SHH | 475.559 | −1.575 |
| FOXN1 | forkhead box N1 | HF development, HS differentiation | WNT, BMP, SHH | 1199.807 | −1.476 |
| FZD2 | frizzled class receptor 2 | receptor WNT pathway | WNT | 155.689 | −0.948 |
| FZD3 | frizzled class receptor 3 | receptor WNT pathway | WNT | 412.223 | −0.978 |
| GLI2 | GLI family zinc finger 2 | HF SC related transcription factor | SHH | 541.121 | −0.927 |
| HHIP | hedgehog interacting protein | HF organogenesis | SHH | 185.270 | −2.185 |
| HOXC13 | homeobox C13 | HS differentiation | WNT | 1064.553 | −1.734 |
| JAG1 | Jagged 1 | HF maintenance | Notch | 5310.336 | −0.668 |
| LEF1 | lymphoid enhancer binding factor 1 | HS differentiation | WNT | 957.142 | −1.636 |
| LGR4 | leucine rich repeat containing G protein-coupled receptor 4 | delays HC; inhibits activation of follicular SCs | WNT | 4786.889 | −0.464 |
| LGR5 | leucine rich repeat containing G protein-coupled receptor 5 | follicular SC marker; anagen initiation | WNT | 1028.425 | −1.430 |
| LGR6 | leucine rich repeat containing G protein-coupled receptor 6 | SC associated marker | WNT | 336.586 | 1.145 |
| LHX2 | LIM homeobox 2 | HF differentiation, SC associated marker | WNT | 645.982 | −1.377 |
| MSX2 | Msh homeobox 2 | HS differentiation | BMP | 295.450 | −1.454 |
| NCAM1 | neural cell adhesion molecule 1 | expressed in dermal papilla | FGF | 267.400 | −1.642 |
| NFATC2IP | nuclear factor of activated T cells 2 interacting protein | aging of HF stem cells | 1056.715 | 0.332 | |
| SHH | Sonic hedgehog | HF development and cycling | SHH | 51.377 | −2.002 |
| SMO | Smoothened | HF development and cycling | SHH | 781.216 | −0.858 |
| Gene Symbol | Full Gene Name | Function | BaseMean | Log2FC |
|---|---|---|---|---|
| CYP27B1 | cytochrome P450 family 27 subfamily B member 1 | activates vitamin D3 | 209.288 | −1.650 |
| CYP2R1 | cytochrome P450 family 2 subfamily R1 | major vitamin D25-hydroxylase | 228.196 | 0.980 |
| CYP39A1 | Cytochrome P450 Family 39 Subfamily A Member 1 | 7-alpha hydroxylation of 24-hydroxycholesterol | 753.872 | −0.818 |
| CYP51A1 | cytochrome P450 family 51 subfamily A member 1 | cholesterol biosynthesis | 1933.523 | 0.712 |
| DHCR7 | 7-Dehydrocholesterol reductase | converts 7-dehydrocholesterol (substrate for vitamin D formation cholesterol) | 2100.759 | 0.719 |
| ESR2 | estrogen receptor 2 | nuclear receptor, expressed in the HF in outer root sheath, dermal papilla, matrix cells and in the bulge | 180.354 | −1.212 |
| HSD17B2 | 17β-Hydroxysteroid dehydrogenase 2 | inactivation of estrogens and androgens: converts estradiol to estrone, testosterone to androstenedione, and androstenediol to DHEA; activates the weak progestogen 20α-hydroxyprogesterone into the potent progestogen progesterone | 2720.505 | 1.061 |
| HSD17B6 | 17β-Hydroxysteroid dehydrogenase 6 | androgen catabolism: convert 3 alpha-adiol to dihydrotestosterone and androsterone to epi-androsterone. | 310.914 | 0.762 |
| HSD17B7 | 17β-Hydroxysteroid dehydrogenase 7 | biosynthesis of estrogen and cholesterol | 1341.347 | 0.687 |
| HSD3B2 | hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 2 | biosynthesis of all classes of hormonal steroids | 154.717 | 2.406 |
| RXRG | retinoid X receptor gamma | increases transcriptional function of VDR | 147.075 | −1.132 |
| VDR | vitamin D receptor | nuclear transcription factor, absence leads to defects in HF regeneration and alopecia | 3347.023 | −1.211 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neradilová, S.; Schauer, A.M.; Hayward, J.J.; Brunner, M.A.T.; Bohutínská, M.; Jagannathan, V.; Connell, L.B.; Boyko, A.R.; Welle, M.M.; Černá Bolfíková, B. Genomic and Transcriptomic Characterization of Atypical Recurrent Flank Alopecia in the Cesky Fousek. Genes 2022, 13, 650. https://doi.org/10.3390/genes13040650
Neradilová S, Schauer AM, Hayward JJ, Brunner MAT, Bohutínská M, Jagannathan V, Connell LB, Boyko AR, Welle MM, Černá Bolfíková B. Genomic and Transcriptomic Characterization of Atypical Recurrent Flank Alopecia in the Cesky Fousek. Genes. 2022; 13(4):650. https://doi.org/10.3390/genes13040650
Chicago/Turabian StyleNeradilová, Silvie, Alexandria M. Schauer, Jessica J. Hayward, Magdalena A. T. Brunner, Magdalena Bohutínská, Vidhya Jagannathan, Laurie B. Connell, Adam R. Boyko, Monika M. Welle, and Barbora Černá Bolfíková. 2022. "Genomic and Transcriptomic Characterization of Atypical Recurrent Flank Alopecia in the Cesky Fousek" Genes 13, no. 4: 650. https://doi.org/10.3390/genes13040650
APA StyleNeradilová, S., Schauer, A. M., Hayward, J. J., Brunner, M. A. T., Bohutínská, M., Jagannathan, V., Connell, L. B., Boyko, A. R., Welle, M. M., & Černá Bolfíková, B. (2022). Genomic and Transcriptomic Characterization of Atypical Recurrent Flank Alopecia in the Cesky Fousek. Genes, 13(4), 650. https://doi.org/10.3390/genes13040650