
scInTime: A Computational Method Leveraging Single-Cell Trajectory and Gene Regulatory Networks to Identify Master Regulators of Cellular Differentiation

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Sets
2.2. Pseudotime Determination for Cells
2.3. Time-Series scGRN Construction and Generate the Regression Coefficient Matrix
2.4. Code Availability
3. Results
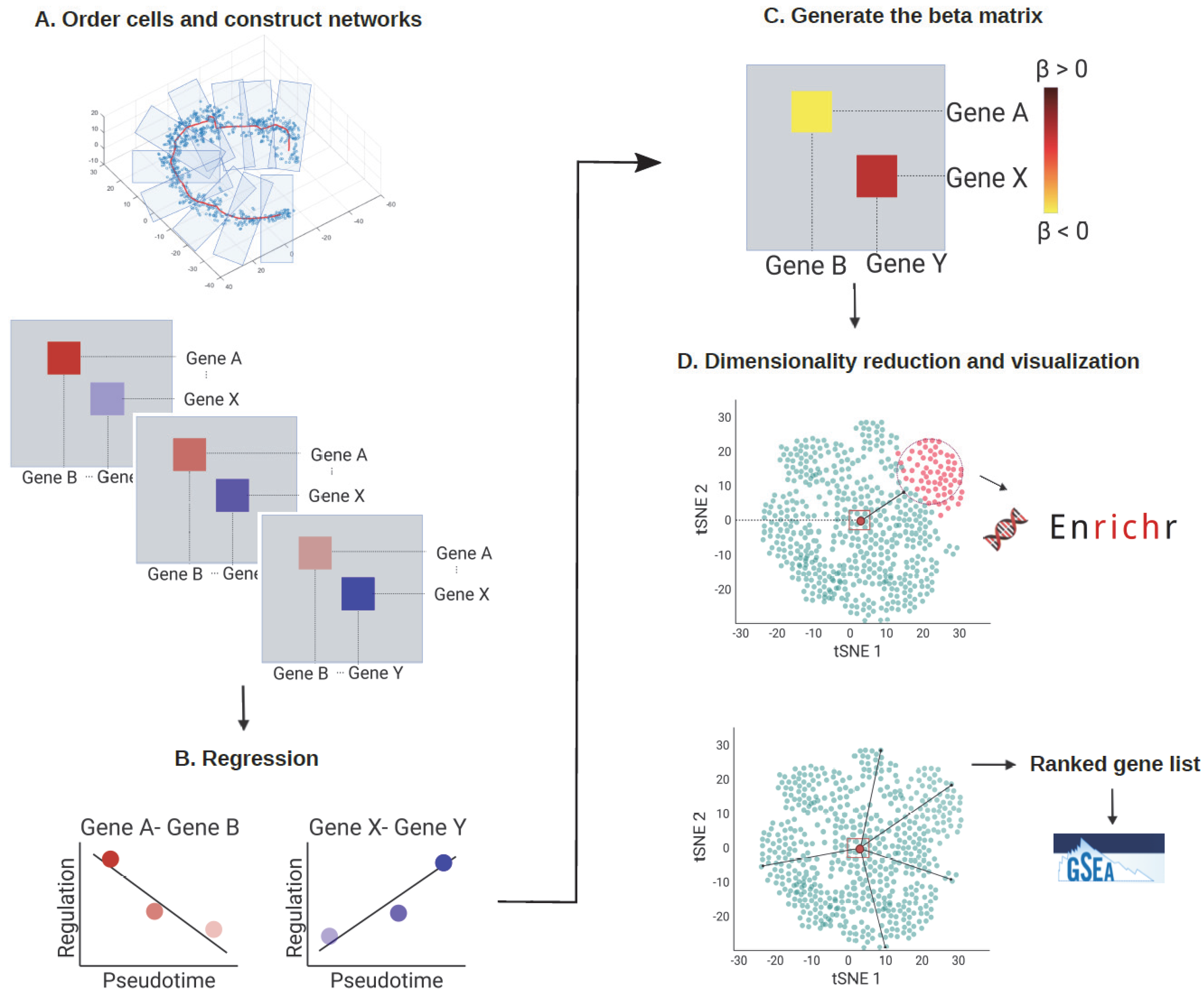
3.1. The scInTime Architecture
3.1.1. Construct Pseudotime-Series Gene Regulatory Networks
3.1.2. Regression Analysis
3.1.3. Build Regression Coefficients Matrix
3.1.4. Analysis of Regression Coefficients Matrix
3.2. Applications to Time-Resolved scRNA-seq Data
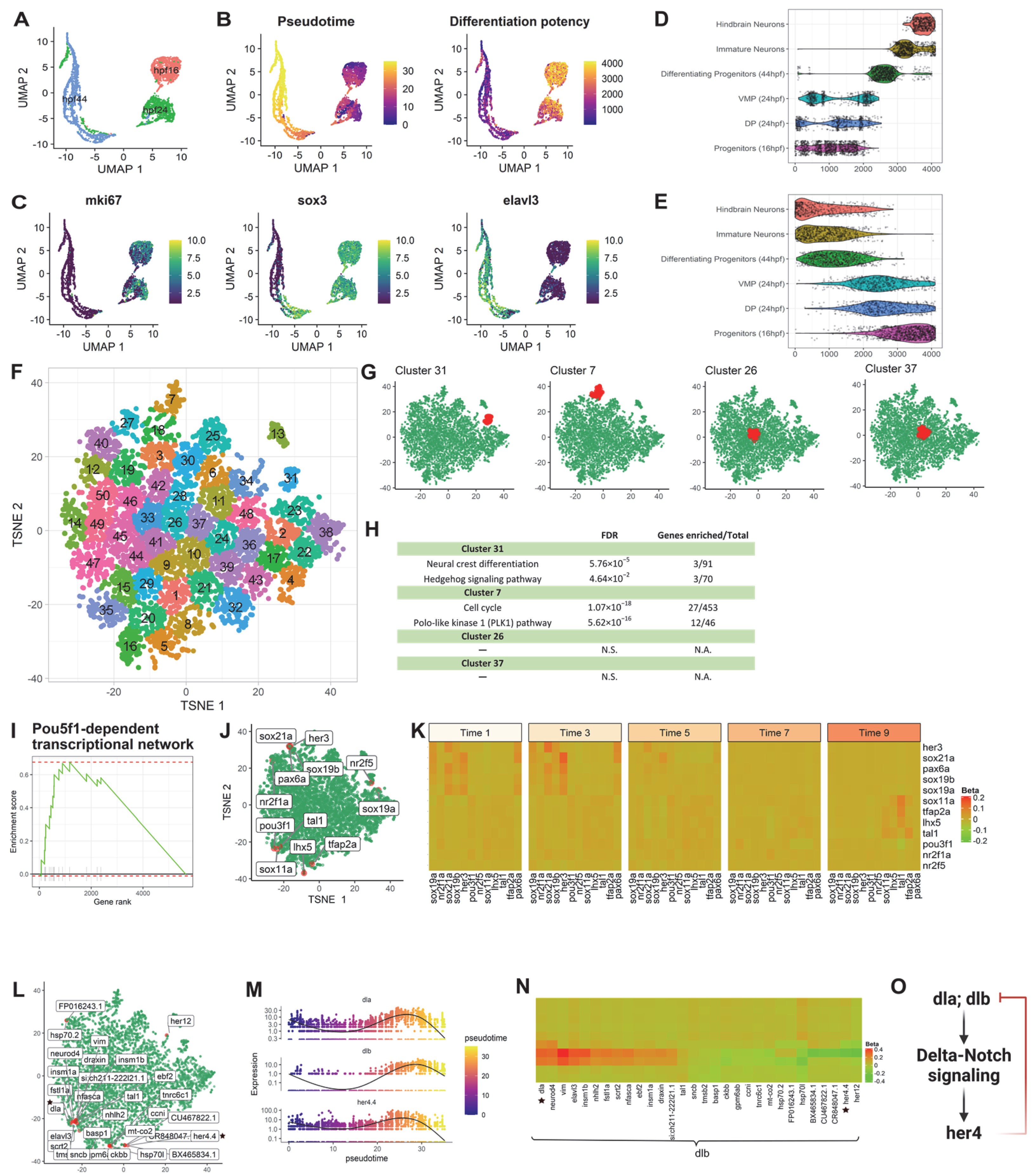
3.2.1. Application 1: Zebrafish Hindbrain
3.2.2. Application 2: HNSCC Cell Line
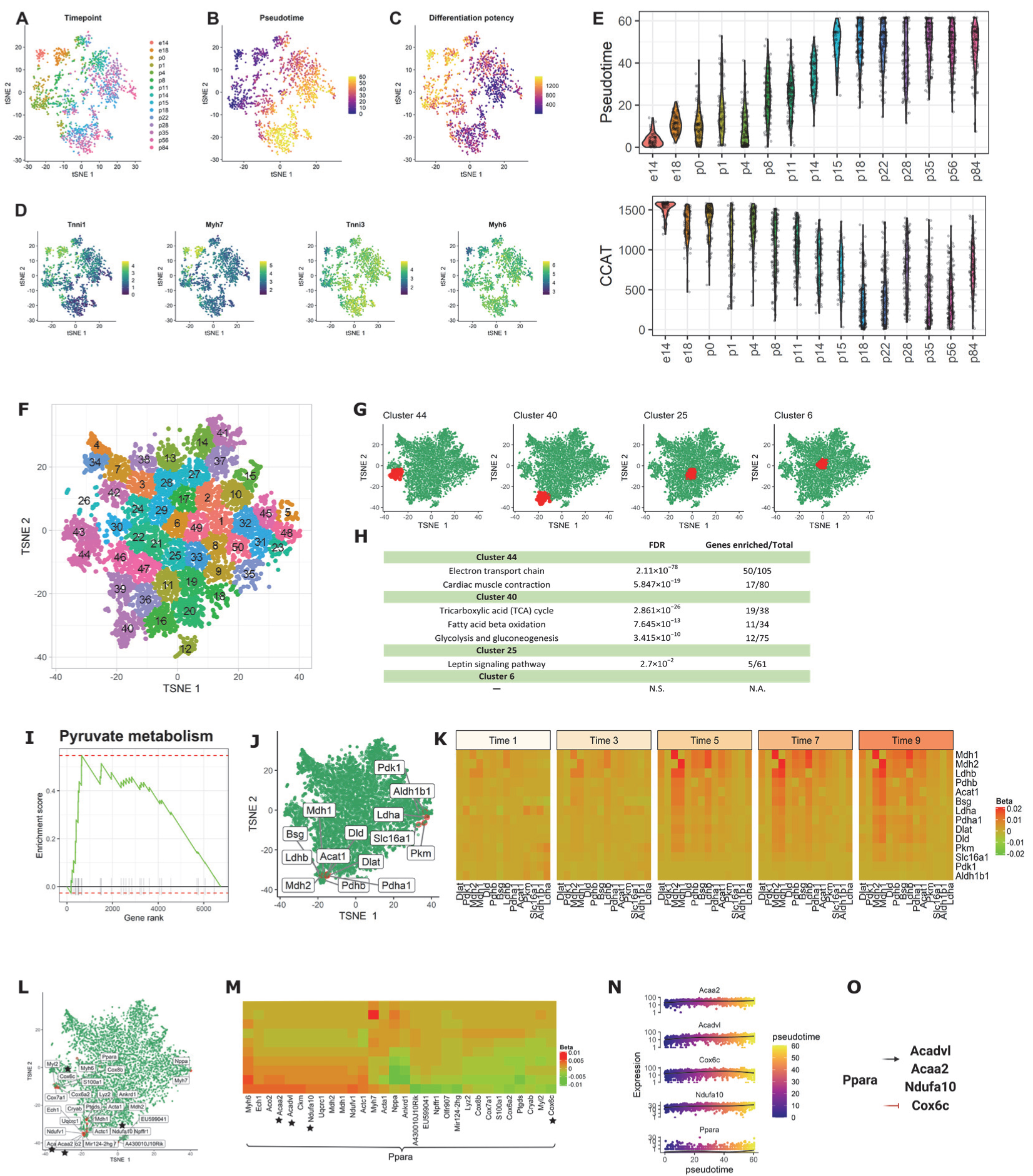
3.2.3. Application 3: Mouse Cardiomyocytes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saelens, W.; Cannoodt, R.; Todorov, H.; Saeys, Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 2019, 37, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.A.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Ji, H. TSCAN: Pseudo-time reconstruction and evaluation in single-cell RNA-seq analysis. Nucleic Acids Res. 2016, 44, e117. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef]
- Lönnberg, T.; Svensson, V.; James, K.R.; Fernandez-Ruiz, D.; Sebina, I.; Montandon, R.; Soon, M.S.F.; Fogg, L.G.; Nair, A.S.; Liligeto, U.N.; et al. Single-cell RNA-seq and computational analysis using temporal mixture modelling resolves Th1/Tfh fate bifurcation in malaria. Sci. Immunol. 2017, 2, eaal2192. [Google Scholar] [CrossRef]
- Van den Berge, K.; de Bézieux, H.R.; Street, K.; Saelens, W.; Cannoodt, R.; Saeys, Y.; Dudoit, S.; Clement, L. Trajectory-based differential expression analysis for single-cell sequencing data. Nat. Commun. 2020, 11, 1201. [Google Scholar] [CrossRef]
- Tambalo, M.; Mitter, R.; Wilkinson, D.G. A single cell transcriptome atlas of the developing zebrafish hindbrain. Development 2020, 147, dev184143. [Google Scholar] [CrossRef]
- Kagohara, L.T.; Zamuner, F.; Davis-Marcisak, E.F.; Sharma, G.; Considine, M.; Allen, J.; Yegnasubramanian, S.; Gaykalova, D.A.; Fertig, E.J. Integrated single-cell and bulk gene expression and ATAC-seq reveals heterogeneity and early changes in pathways associated with resistance to cetuximab in HNSCC-sensitive cell lines. Br. J. Cancer 2020, 123, 101–113. [Google Scholar] [CrossRef]
- Kannan, S.; Miyamoto, M.; Lin, B.L.; Kwon, C. Trajectory reconstruction identifies dysregulation of perinatal maturation programs in pluripotent stem cell-derived cardiomyocytes. bioRxiv 2021. 2021.01.31.428969. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., III; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.E21. [Google Scholar] [CrossRef]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Teschendorff, A.E.; Maity, A.K.; Hu, X.; Weiyan, C.; Lechner, M. Ultra-fast scalable estimation of single-cell differentiation potency from scRNA-Seq data. Bioinformatics 2021, 37, 1528–1534. [Google Scholar] [CrossRef]
- Lause, J.; Berens, P.; Kobak, D. Analytic Pearson residuals for normalization of single-cell RNA-seq UMI data. Genome Biol. 2021, 22, 1–20. [Google Scholar] [CrossRef]
- Osorio, D.; Zhong, Y.; Li, G.; Huang, J.Z.; Cai, J.J. scTenifoldNet: A Machine Learning Workflow for Constructing and Comparing Transcriptome-wide Gene Regulatory Networks from Single-Cell Data. Gene Expr. Patterns 2020, 1, 100139. [Google Scholar] [CrossRef]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Moon, K.R.; van Dijk, D.; Wang, Z.; Gigante, S.; Burkhardt, D.B.; Chen, W.S.; Yim, K.; Elzen, A.V.D.; Hirn, M.J.; Coifman, R.R.; et al. Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol. 2019, 37, 1482–1492. [Google Scholar] [CrossRef]
- Onichtchouk, D.; Geier, F.; Polok, B.; Messerschmidt, D.M.; Mössner, R.; Wendik, B.; Song, S.; Taylor, V.; Timmer, J.; Driever, W. Zebrafish Pou5f1-dependent transcriptional networks in temporal control of early development. Mol. Syst. Biol. 2010, 6, 354. [Google Scholar] [CrossRef]
- Okuda, Y.; Yoda, H.; Uchikawa, M.; Furutani-Seiki, M.; Takeda, H.; Kondoh, H.; Kamachi, Y. Comparative genomic and expression analysis of group B1 sox genes in zebrafish indicates their diversifi-cation during vertebrate evolution. Dev. Dyn. 2006, 235, 811–825. [Google Scholar] [CrossRef]
- Li, X.; Zhou, W.; Li, X.; Gao, M.; Ji, S.; Tian, W.; Ji, G.; Du, J.; Hao, A. SOX19b regulates the premature neuronal differentiation of neural stem cells through EZH2-mediated histone methylation in neural tube development of zebrafish. Stem Cell Res. Ther. 2019, 10, 389. [Google Scholar] [CrossRef] [PubMed]
- Appel, B.; Givan, L.A.; Eisen, J.S. Delta-Notch signaling and lateral inhibition in zebrafish spinal cord development. BMC Dev. Biol. 2001, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.G.; Wen, W.; Pillai-Kastoori, L.; Morris, A.C. Tracking the fate of her4 expressing cells in the regenerating retina using her4:Kaede zebrafish. Exp. Eye Res. 2015, 145, 75–87. [Google Scholar] [CrossRef]
- Radosevic, M.; Fargas, L.; Alsina, B. The Role of her4 in Inner Ear Development and Its Relationship with Proneural Genes and Notch Signalling. PLoS ONE 2014, 9, e109860. [Google Scholar] [CrossRef] [PubMed]
- Kneissl, J.; Keller, S.; Lorber, T.; Heindl, S.; Keller, G.; Drexler, I.; Hapfelmeier, A.; Höfler, H.; Luber, B. Association of amphiregulin with the cetuximab sensitivity of gastric cancer cell lines. Int. J. Oncol. 2012, 41, 733–744. [Google Scholar] [CrossRef][Green Version]
- Hübner, J.; Raschke, M.; Rütschle, I.; Gräßle, S.; Hasenberg, T.; Schirrmann, K.; Lorenz, A.; Schnurre, S.; Lauster, R.; Maschmeyer, I.; et al. Simultaneous evaluation of anti-EGFR-induced tumour and adverse skin effects in a microfluidic human 3D co-culture model. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef]
- Sainio, A.; Järveläinen, H. Extracellular matrix-cell interactions: Focus on therapeutic applications. Cell. Signal. 2020, 66, 109487. [Google Scholar] [CrossRef]
- Zhang, X.; Song, Y.; Song, N.; Zhang, Y.; Zhang, L.; Wang, Y.; Wang, Z.-N.; Qu, X.; Liu, Y. RANKL/RANK pathway abrogates cetuximab sensitivity in gastric cancer cells via activation of EGFR and c-Src. OncoTargets Ther. 2017, 10, 73–83. [Google Scholar] [CrossRef]
- Holcakova, J.; Nekulova, M.; Orzol, P.; Nenutil, R.; Podhorec, J.; Svoboda, M.; Dvorakova, P.; Pjechova, M.; Hernychova, L.; Vojtesek, B.; et al. ΔNp63 activates EGFR signaling to induce loss of adhesion in triple-negative basal-like breast cancer cells. Breast Cancer Res. Treat. 2017, 163, 475–484. [Google Scholar] [CrossRef]
- Compagnone, M.; Gatti, V.; Presutti, D.; Ruberti, G.; Fierro, C.; Markert, E.K.; Vousden, K.H.; Zhou, H.; Mauriello, A.; Anemone, L.; et al. ΔNp63-mediated regulation of hyaluronic acid metabolism and signaling supports HNSCC tu-morigenesis. Proc. Natl. Acad. Sci. USA 2017, 114, 13254–13259. [Google Scholar] [CrossRef] [PubMed]
- Sundqvist, A.; Vasilaki, E.; Voytyuk, O.; Bai, Y.; Morikawa, M.; Moustakas, A.; Miyazono, K.; Heldin, C.H.; ten Dijke, P.; van Dam, H. TGFβ and EGF signaling orchestrates the AP-1- and p63 transcriptional regulation of breast cancer in-vasiveness. Oncogene 2020, 39, 4436–4449. [Google Scholar] [CrossRef] [PubMed]
- Scartozzi, M.; Bearzi, I.; Pierantoni, C.; Mandolesi, A.; Loupakis, F.; Zaniboni, A.; Catalano, V.; Quadri, A.; Zorzi, F.; Berardi, R.; et al. Nuclear factor-kB tumor expression predicts response and survival in irinotecan-refractory metastatic colorectal cancer treated with cetuximab-irinotecan therapy. J. Clin. Oncol. 2007, 25, 3930–3935. [Google Scholar] [CrossRef]
- Fouani, L.; Kovacevic, Z.; Richardson, D.R. Targeting Oncogenic Nuclear Factor Kappa B Signaling with Redox-Active Agents for Cancer Treatment. Antioxid. Redox Signal. 2019, 30, 1096–1123. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Park, S.Y.; Kang, K.B.; Park, R.W.; Kim, I.S. NF-kappaB activates fibronectin gene expression in rat hepatocytes. Biochem. Biophys. Res. Commun. 2002, 297, 1218–1224. [Google Scholar] [CrossRef]
- Zaidi, A.H.; Manna, S.K. Profilin-PTEN interaction suppresses NF-κB activation via inhibition of IKK phosphorylation. Biochem. J. 2016, 473, 859–872. [Google Scholar] [CrossRef]
- Pan, Y.; Huang, J.; Xing, R.; Yin, X.; Cui, J.; Li, W.; Yu, J.; Lu, Y. Metallothionein 2A inhibits NF-κB pathway activation and predicts clinical outcome segregated with TNM stage in gastric cancer patients following radical resection. J. Transl. Med. 2013, 11, 173. [Google Scholar] [CrossRef]
- Lin, S.; Wang, X.; Pan, Y.; Tian, R.; Lin, B.; Jiang, G.-S.; Chen, K.; He, Y.-Q.; Zhang, L.; Zhai, W.; et al. Transcription Factor Myeloid Zinc-Finger 1 Suppresses Human Gastric Carcinogenesis by Interacting with Metallothionein 2A. Clin. Cancer Res. 2019, 25, 1050–1062. [Google Scholar] [CrossRef]
- Shi, X.; Fan, C.; Jiu, Y. Unidirectional Regulation of Vimentin Intermediate Filaments to Caveolin-1. Int. J. Mol. Sci. 2020, 21, 7436. [Google Scholar] [CrossRef]
- Guo, Y.; Pu, W.T. Cardiomyocyte Maturation: New Phase in Development. Circ. Res. 2020, 126, 1086–1106. [Google Scholar] [CrossRef]
- Cui, Y.; Zheng, Y.; Liu, X.; Yan, L.; Fan, X.; Yong, J.; Hu, Y.; Dong, J.; Li, Q.; Wu, X.; et al. Single-Cell Transcriptome Analysis Maps the Developmental Track of the Human Heart. Cell Rep. 2019, 26, 1934–1950.e5. [Google Scholar] [CrossRef] [PubMed]
- Reiser, P.J.; Portman, M.A.; Ning, X.-H.; Moravec, C.S. Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am. J. Physiol. Circ. Physiol. 2001, 280, H1814–H1820. [Google Scholar] [CrossRef] [PubMed]
- Bedada, F.B.; Chan, S.S.-K.; Metzger, S.K.; Zhang, L.; Zhang, J.; Garry, D.J.; Kamp, T.; Kyba, M.; Metzger, J.M. Acquisition of a Quantitative, Stoichiometrically Conserved Ratiometric Marker of Maturation Status in Stem Cell-Derived Cardiac Myocytes. Stem Cell Rep. 2014, 3, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Dzeja, P.P.; Faustino, R.S.; Perez-Terzic, C.; Behfar, A.; Terzic, A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, S60–S67. [Google Scholar] [CrossRef]
- Morita, Y.; Tohyama, S. Metabolic Regulation of Cardiac Differentiation and Maturation in Pluripotent Stem Cells: A Lesson from Heart Development. JMA J. 2020, 3, 193–200. [Google Scholar] [CrossRef]
- Malandraki-Miller, S.; Lopez, C.A.; Al-Siddiqi, H.; Carr, C.A. Changing Metabolism in Differentiating Cardiac Progenitor Cells—Can Stem Cells Become Metabolically Flexible Cardiomyocytes? Front. Cardiovasc. Med. 2018, 5, 119. [Google Scholar] [CrossRef]
- Murphy, S.A.; Miyamoto, M.; Kervadec, A.; Kannan, S.; Tampakakis, E.; Kambhampati, S.; Lin, B.L.; Paek, S.; Andersen, P.; Lee, D.I.; et al. PGC1/PPAR drive cardiomyocyte maturation at single cell level via YAP1 and SF3B2. Nat. Commun. 2021, 12, 1648. [Google Scholar] [CrossRef]
- Auboeuf, D.; Rieusset, J.; Fajas, L.; Vallier, P.; Frering, V.; Riou, J.P.; Staels, B.; Auwerx, J.; Laville, M.; Vidal, H. Tissue Distribution and Quantification of the Expression of mRNAs of Peroxisome Proliferator—Activated Receptors and Liver X Receptor-α in Humans: No Alteration in Adipose Tissue of Obese and NIDDM Patients. Diabetes 1997, 46, 1319–1327. [Google Scholar] [CrossRef]
- Inoue, I.; Shino, K.; Noji, S.; Awata, T.; Katayama, S. Expression of peroxisome proliferator-activated receptor alpha (PPAR alpha) in primary cultures of human vascular endothelial cells. Biochem. Biophys. Res. Commun. 1998, 246, 370–374. [Google Scholar] [CrossRef]
- Wang, G.; Namura, S. Effects of chronic systemic treatment with peroxisome proliferator-activated receptor α activators on neuroinflammation induced by intracerebral injection of lipopolysaccharide in adult mice. Neurosci. Res. 2011, 70, 230–237. [Google Scholar] [CrossRef]
- Santin, J.R.; Uchôa, F.D.; Maria do Carmo, A.L.; Rabello, M.M.; Machado, I.D.; Hernandes, M.Z.; Amato, A.A.; Milton, F.A.; Webb, P.; Neves, F.D.A.R.; et al. Chemical synthesis, docking studies and biological effects of a pan peroxisome proliferator-activated re-ceptor agonist and cyclooxygenase inhibitor. Eur. J. Pharm. Sci. 2013, 48, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Combs, C.K.; Bates, P.; Karlo, J.C.; Landreth, G.E. Regulation of beta-amyloid stimulated proinflammatory responses by peroxisome proliferator-activated receptor alpha. Neurochem. Int. 2001, 39, 449–457. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Q.; Li, G.; Osorio, D.; Zhong, Y.; Yang, Y.; Lin, Y.-T.; Zhang, X.; Cai, J.J. scInTime: A Computational Method Leveraging Single-Cell Trajectory and Gene Regulatory Networks to Identify Master Regulators of Cellular Differentiation. Genes 2022, 13, 371. https://doi.org/10.3390/genes13020371
Xu Q, Li G, Osorio D, Zhong Y, Yang Y, Lin Y-T, Zhang X, Cai JJ. scInTime: A Computational Method Leveraging Single-Cell Trajectory and Gene Regulatory Networks to Identify Master Regulators of Cellular Differentiation. Genes. 2022; 13(2):371. https://doi.org/10.3390/genes13020371
Chicago/Turabian StyleXu, Qian, Guanxun Li, Daniel Osorio, Yan Zhong, Yongjian Yang, Yu-Te Lin, Xiuren Zhang, and James J. Cai. 2022. "scInTime: A Computational Method Leveraging Single-Cell Trajectory and Gene Regulatory Networks to Identify Master Regulators of Cellular Differentiation" Genes 13, no. 2: 371. https://doi.org/10.3390/genes13020371
APA StyleXu, Q., Li, G., Osorio, D., Zhong, Y., Yang, Y., Lin, Y.-T., Zhang, X., & Cai, J. J. (2022). scInTime: A Computational Method Leveraging Single-Cell Trajectory and Gene Regulatory Networks to Identify Master Regulators of Cellular Differentiation. Genes, 13(2), 371. https://doi.org/10.3390/genes13020371