
A Genome-Scale Metabolic Model for the Human Pathogen Candida Parapsilosis and Early Identification of Putative Novel Antifungal Drug Targets


 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Model Construction
2.1.1. Enzyme and Reaction Annotation
2.1.2. Correcting Reaction Reversibility, Directionality, and Balance
2.1.3. Compartmentalisation
2.1.4. Defining the Biomass Equation
2.2. Model Simulations and Enzyme Essentiality Prediction
2.3. Model Validation
2.3.1. Strains and Growth Media
2.3.2. Aerobic Batch Cultivation
2.3.3. Cell Density, Dry Weight, and Metabolite Concentration Assessment
3. Results and Discussion
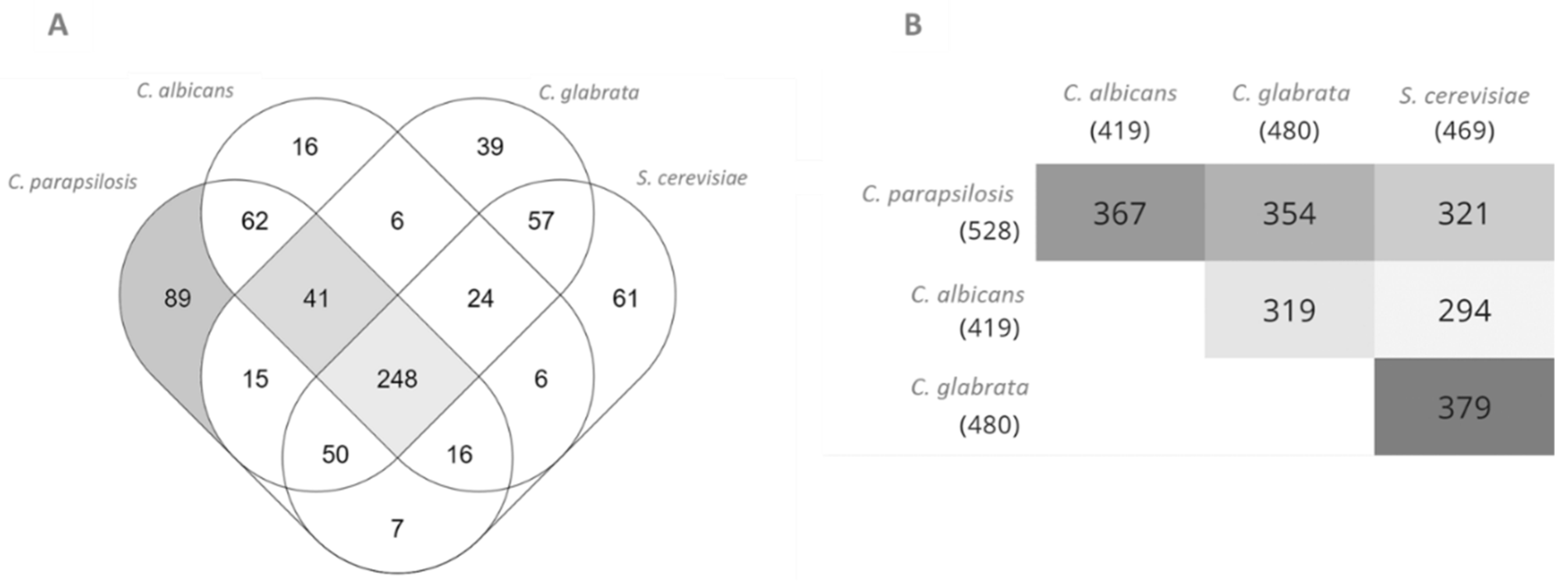
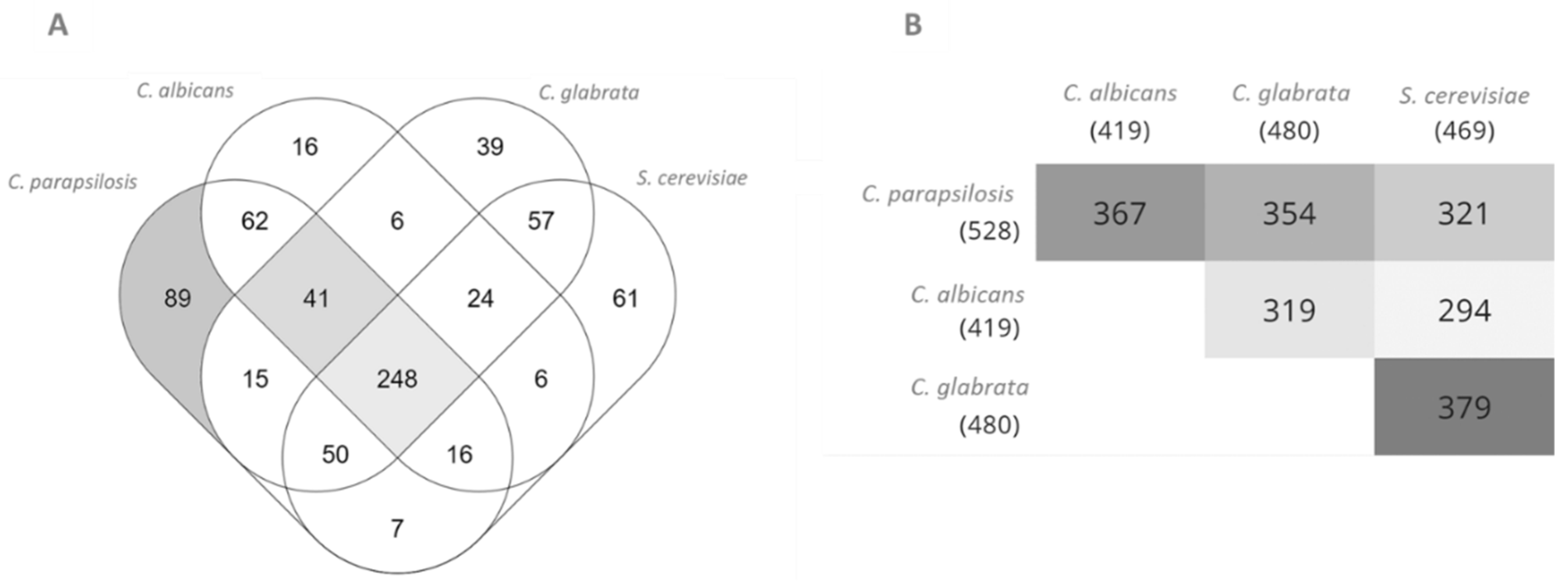
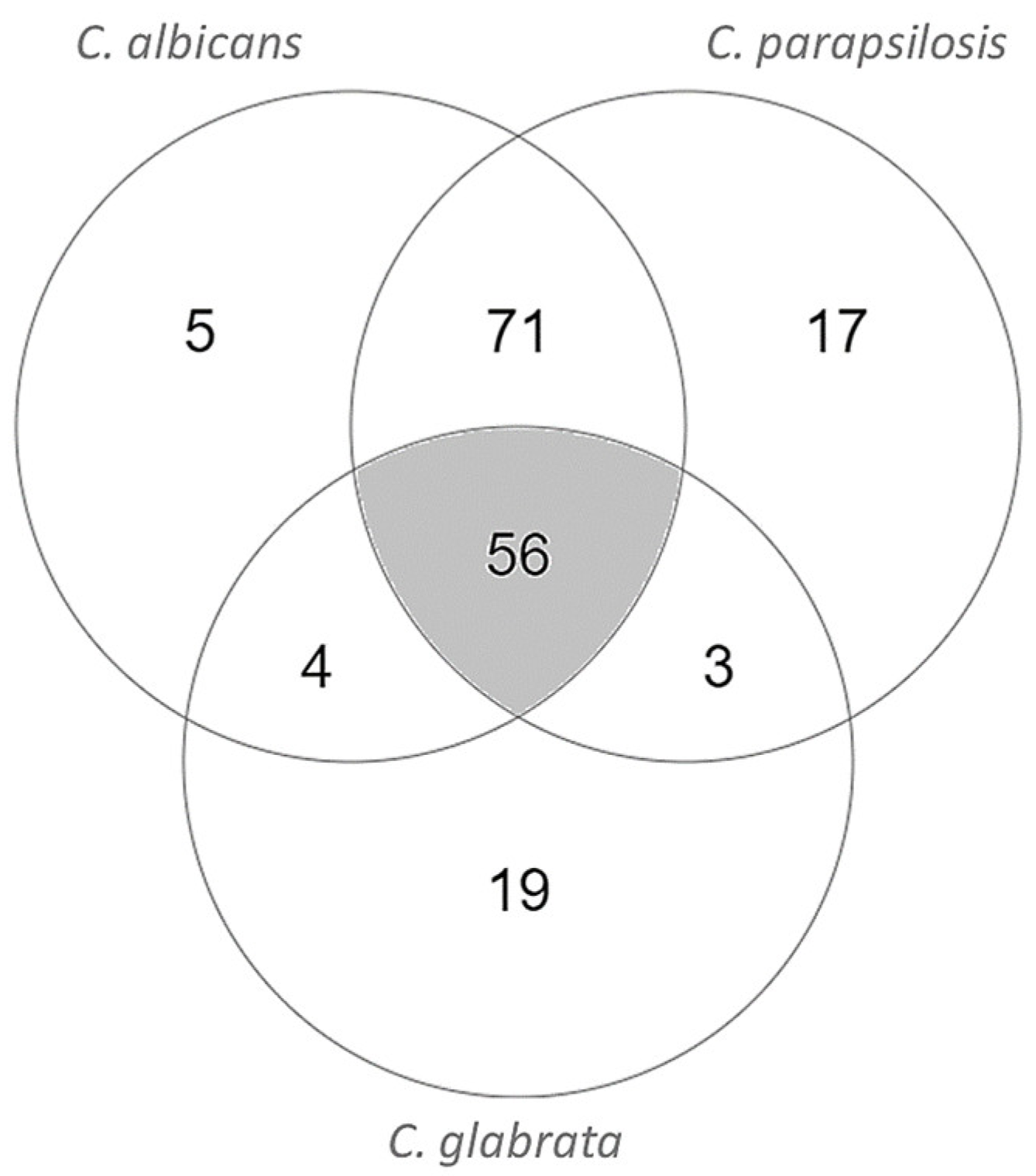
3.1. Model Characteristics, Highlighting C. Parapsilosis Unique Metabolic Features
3.2. Model Validation
3.2.1. Assessing the Model’s Ability to Predict Carbon and Nitrogen Source Usage
3.2.2. Assessing the Model’s Ability to Quantitatively Predict Growth Parameters
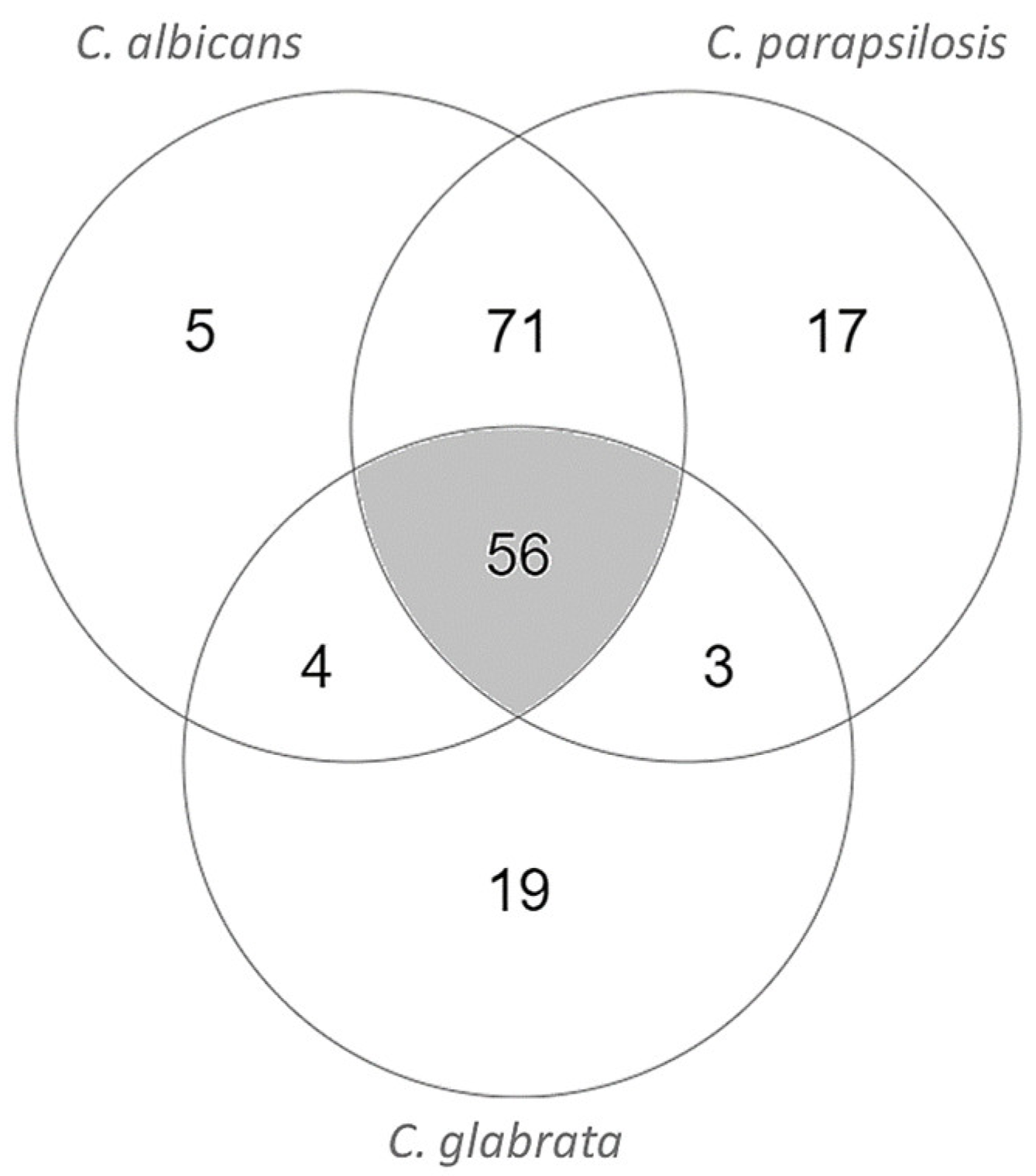
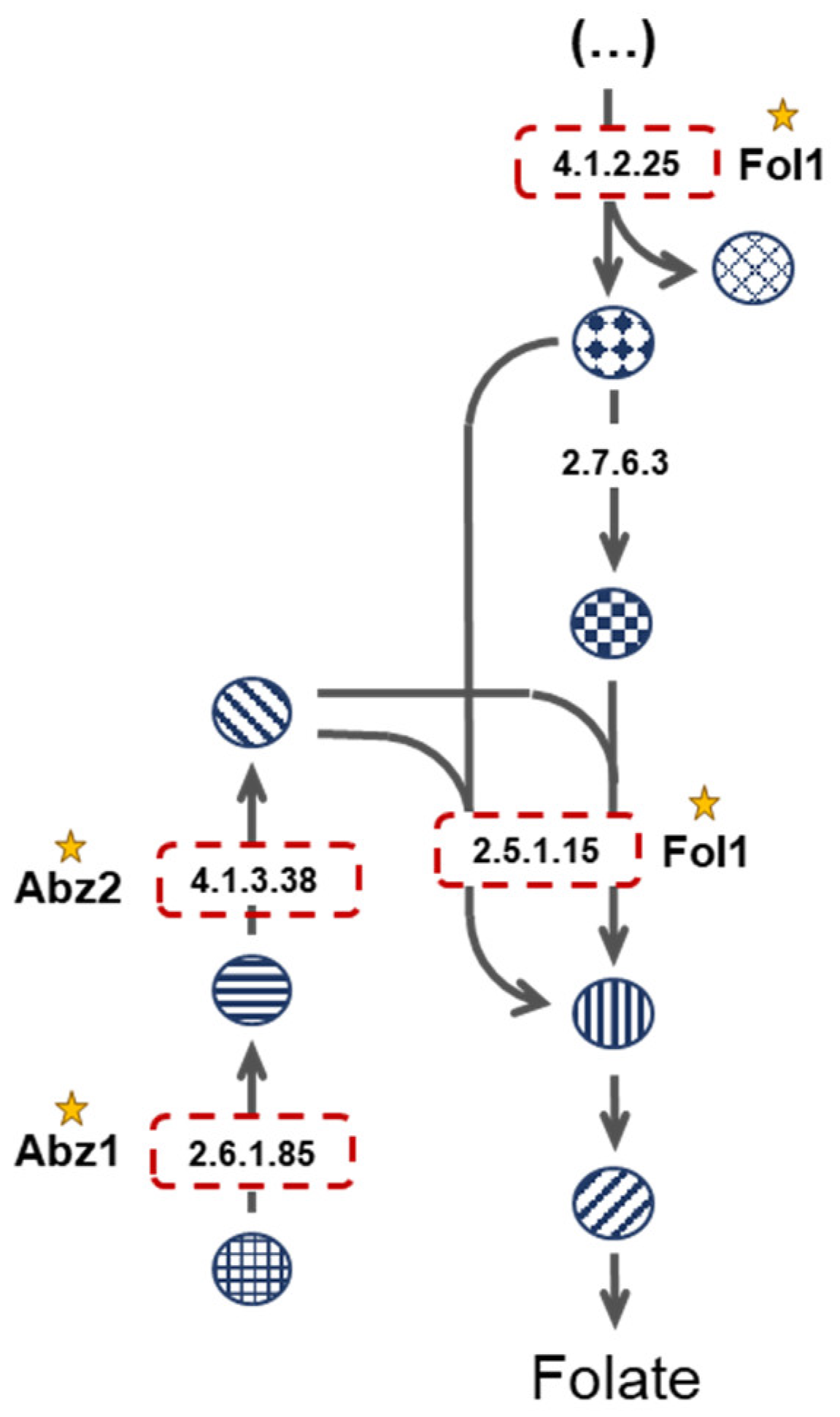
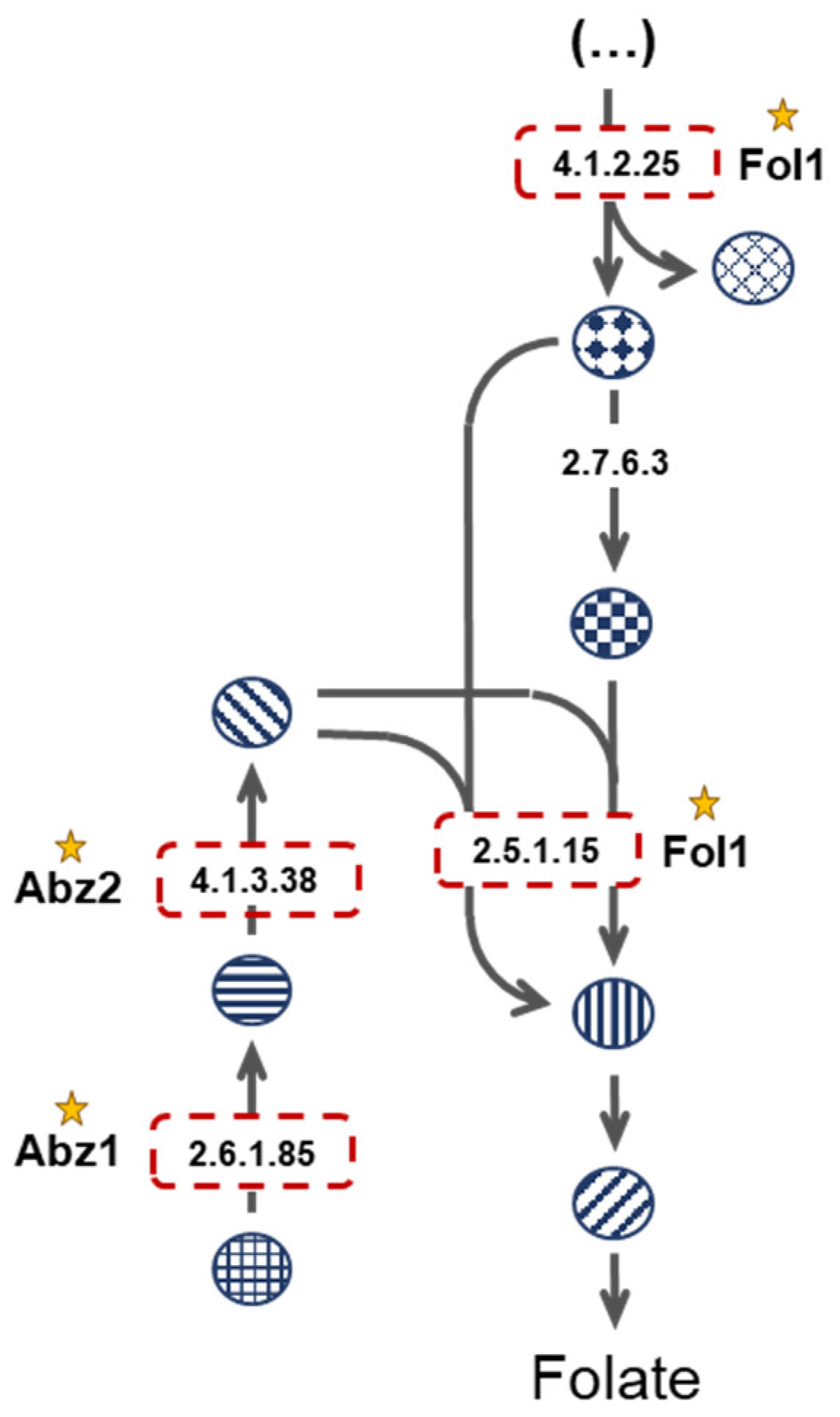
3.3. Enzyme Essentiality Assessment: Looking for New Drug Targets
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Silva, S.; Negri, M.; Henriques, M.; Oliveira, R.; Williams, D.W.; Azeredo, J. Candida glabrata, Candida parapsilosis and Candida tropicalis: Biology, epidemiology, pathogenicity and antifungal resistance. FEMS Microbiol. Rev. 2012, 36, 288–305. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, R.A.; Fridkin, S.K. The Changing Face of Fungal Infections in Health Care Settings. Clin. Infect. Dis. 2005, 41, 1455–1460. [Google Scholar] [CrossRef]
- Trofa, D.; Gácser, A.; Nosanchuk, J.D. Candida parapsilosis, an Emerging Fungal Pathogen. Clin. Microbiol. Rev. 2008, 21, 606–625. [Google Scholar] [CrossRef] [Green Version]
- Castanheira, M.; Deshpande, L.M.; Messer, S.A.; Rhomberg, P.R.; Pfaller, M.A. Analysis of global antifungal surveillance results reveals predominance of Erg11 Y132F alteration among azole-resistant Candida parapsilosis and Candida tropicalis and country-specific isolate dissemination. Int. J. Antimicrob. Agents 2020, 55, 105799. [Google Scholar] [CrossRef]
- Gu, C.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current status and applications of genome-scale metabolic models. Genome Biol. 2019, 20, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Hua, Q. Applications of Genome-Scale Metabolic Models in Biotechnology and Systems Medicine. Front. Physiol. 2016, 6, 413. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.S.; Palsson, B.O. Systems Properties of the Haemophilus influenzaeRd Metabolic Genotype. J. Biol. Chem. 1999, 274, 17410–17416. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.Y.; Kim, H.U.; Lee, S.Y. Metabolite-centric approaches for the discovery of antibacterials using genome-scale metabolic networks. Metab. Eng. 2010, 12, 105–111. [Google Scholar] [CrossRef]
- Dias, O.; Pereira, R.; Gombert, A.K.; Ferreira, E.C.; Rocha, I. iOD907, the first genome-scale metabolic model for the milk yeast Kluyveromyces lactis. Biotechnol. J. 2014, 9, 776–790. [Google Scholar] [CrossRef] [Green Version]
- Raškevičius, V.; Mikalayeva, V.; Antanavičiūtė, I.; Ceslevičienė, I.; Skeberdis, V.A.; Kairys, V.; Bordel, S. Genome scale metabolic models as tools for drug design and personalized medicine. PLoS ONE 2018, 13, e0190636. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.L.; Nielsen, J. Anticancer drug discovery through genome-scale metabolic modeling. Curr. Opin. Syst. Biol. 2017, 4, 1–8. [Google Scholar] [CrossRef]
- Mienda, B.S.; Salihu, R.; Adamu, A.; Idris, S. Genome-scale metabolic models as platforms for identification of novel genes as antimicrobial drug targets. Future Microbiol. 2018, 13, 455–467. [Google Scholar] [CrossRef]
- Dias, O.; Saraiva, J.P.; Faria, C.; Ramirez, M.; Pinto, F.; Rocha, I. iDS372, a Phenotypically Reconciled Model for the Metabolism of Streptococcus pneumoniae Strain R. Front. Microbiol. 2019, 10, 1283. [Google Scholar] [CrossRef]
- Viana, R.; Dias, O.; Lagoa, D.; Galocha, M.; Rocha, I.; Teixeira, M.C. Genome-Scale Metabolic Model of the Human Pathogen Candida albicans: A Promising Platform for Drug Target Prediction. J. Fungi 2020, 6, 171. [Google Scholar] [CrossRef]
- Rocha, I.; Maia, P.; Evangelista, P.; Vilaça, P.; Soares, S.; Pinto, J.P.; Nielsen, J.; Patil, K.R.; Ferreira, E.C.; Rocha, M. OptFlux: An open-source software platform for in silico metabolic engineering. BMC Syst. Biol. 2010, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [Green Version]
- Dias, O.; Rocha, M.; Ferreira, E.C.; Rocha, I. Reconstructing genome-scale metabolic models with merlin. Nucleic Acids Res. 2015, 43, 3899–3910. [Google Scholar] [CrossRef]
- Kitts, P.A.; Church, D.M.; Thibaud-Nissen, F.; Choi, J.; Hem, V.; Sapojnikov, V.; Smith, R.G.; Tatusova, T.A.; Xiang, C.; Zherikov, A.; et al. Assembly: A resource for assembled genomes at NCBI. Nucleic Acids Res. 2016, 44, D73–D80. [Google Scholar] [CrossRef]
- Federhen, S. The NCBI Taxonomy database. Nucleic Acids Res. 2011, 40, D136–D143. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bansal, P.; Bridge, A.J.; Poux, S.; Bougueleret, L.; Xenarios, I. UniProtKB/Swiss-Prot, the Manually Annotated Section of the UniProt KnowledgeBase: How to Use the Entry View. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; pp. 23–54. [Google Scholar] [CrossRef]
- Dias, O.; Rocha, M.; Ferreira, E.C.; Rocha, I. Reconstructing High-Quality Large-Scale Metabolic Models with merlin. In Programmed Necrosis; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2018; Volume 1716, pp. 1–36. [Google Scholar]
- Tsui, C.K.; Daniel, H.-M.; Robert, V.; Meyer, W. Re-examining the phylogeny of clinically relevant Candida species and allied genera based on multigene analyses. FEMS Yeast Res. 2008, 8, 651–659. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Flamholz, A.; Noor, E.; Bar-Even, A.; Milo, R. eQuilibrator—The biochemical thermodynamics calculator. Nucleic Acids Res. 2011, 40, D770–D775. [Google Scholar] [CrossRef] [Green Version]
- Caspi, R.; Altman, T.; Billington, R.; Dreher, K.; Foerster, H.; Fulcher, C.A.; Holland, T.A.; Keseler, I.M.; Kothari, A.; Kubo, A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014, 42, D459–D471. [Google Scholar] [CrossRef] [Green Version]
- Degtyarenko, K.; De Matos, P.; Ennis, M.; Hastings, J.; Zbinden, M.; McNaught, A.; Alcántara, R.; Darsow, M.; Guedj, M.; Ashburner, M. ChEBI: A database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2007, 36, D344–D350. [Google Scholar] [CrossRef]
- Schomburg, D. BRENDA, enzyme data and metabolic information. Nucleic Acids Res. 2002, 30, 47–49. [Google Scholar] [CrossRef]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [Green Version]
- Saier, M.H., Jr.; Reddy, V.S.; Moreno-Hagelsieb, G.; Hendargo, K.J.; Zhang, Y.; Iddamsetty, V.; Lam, K.J.K.; Tian, N.; Russum, S.; Wang, J.; et al. The Transporter Classification Database (TCDB): 2021 update. Nucleic Acids Res. 2021, 49, D461–D467. [Google Scholar] [CrossRef]
- Lagoa, D. Development of Bioinformatics Tools for the Classification of Transporter Systems; University of Minho: Braga, Portugal, 2019. [Google Scholar]
- Santos, S.; Rocha, I. A Computation Tool for the Estimation of Biomass Composition from Genomic and Transcriptomic Information. Adv. Hum. Error Reliab. Resil. Perform. 2016, 13, 161–169. [Google Scholar] [CrossRef]
- Mishra, P.; Park, G.-Y.; Lakshmanan, M.; Lee, H.-S.; Lee, H.; Chang, M.W.; Ching, C.B.; Ahn, J.; Lee, D.-Y. Genome-scale metabolic modeling and in silico analysis of lipid accumulating yeast Candida tropicalis for dicarboxylic acid production. Biotechnol. Bioeng. 2016, 113, 1993–2004. [Google Scholar] [CrossRef]
- Varma, A.; O. Palsson, B. Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110. Appl. Environ. Microbiol. 1994, 60, 3724–3731. [Google Scholar] [CrossRef] [Green Version]
- Sauer, U.; Lasko, D.R.; Fiaux, J.; Hochuli, M.; Glaser, R.; Szyperski, T.; Wüthrich, K.; Bailey, J.E. Metabolic Flux Ratio Analysis of Genetic and Environmental Modulations of Escherichia coli Central Carbon Metabolism. J. Bacteriol. 1999, 181, 6679–6688. [Google Scholar] [CrossRef] [Green Version]
- Pfaller, M.; Riley, J. Effects of fluconazole on the sterol and carbohydrate composition of four species of Candida. Eur. J. Clin. Microbiol. 1992, 11, 152–156. [Google Scholar] [CrossRef]
- Ghannoum, M.A.; Abu-Elteen, K.H. Pathogenicity determinants of Candida. Mycoses 1990, 33, 265–282. [Google Scholar] [CrossRef]
- Mayatepek, E.; Herz, A.; Leichsenring, M.; Kappe, R. Fatty Acid Analysis of Different Candida Species by Capillary Column Gas-Liquid Chromatography. Mycoses 1991, 34, 53–57. [Google Scholar] [CrossRef]
- Thiele, I.; Palsson, B.O. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Liu, L.; Zou, W.; Liu, J.; Hua, Q.; Chen, J. Reconstruction and analysis of the genome-scale metabolic network of Candida glabrata. Mol. BioSyst. 2013, 9, 205–216. [Google Scholar] [CrossRef]
- Mo, M.L.; Palsson, B.Ø.; Herrgård, M.J. Connecting extracellular metabolomic measurements to intracellular flux states in yeast. BMC Syst. Biol. 2009, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Endoh, R.; Horiyama, M.; Ohkuma, M. D-Fructose Assimilation and Fermentation by Yeasts Belonging to Saccharomycetes: Rediscovery of Universal Phenotypes and Elucidation of Fructophilic Behaviors in Ambrosiozyma platypodis and Cyberlindnera americana. Microorganisms 2021, 9, 758. [Google Scholar] [CrossRef]
- Fell, J.W.; Meyer, S.A. Systematics of yeast species in the Candida parapsilosis group. Mycopathol. Mycol. Appl. 1967, 32, 177–193. [Google Scholar] [CrossRef]
- Tambosis, E.; Atkins, B.L.; Capizzi, T.; Gottlieb, T. Rapid and cost-effective identification of Candida species using multipoint inoculation of CHROMagar Candida media, cycloheximide sensitivity and carbohydrate assimilation tests. Pathology 2003, 35, 151–156. [Google Scholar] [CrossRef]
- Devadas, S.M. Auxanographic Carbohydrate Assimilation Method for Large Scale Yeast Identification. J. Clin. Diagn. Res. 2017, 11, DC01–DC03. [Google Scholar] [CrossRef]
- Deorukhkar, S.C.; Roushani, S. Identification of Candida Species: Conventional Methods in the Era of Molecular Diagnosis. Ann. Microbiol. Immunol. 2018, 1, 1002. [Google Scholar]
- CBS-KNAW Collections. Available online: http://www.cbs.knaw.nl/Collections (accessed on 24 July 2020).
- Turner, S.A.; Ma, Q.; Ola, M.; Vicente, K.M.D.S.; Butler, G. Dal81 Regulates Expression of Arginine Metabolism Genes in Candida parapsilosis. mSphere 2018, 3, e00028-18. [Google Scholar] [CrossRef] [Green Version]
- Masaki, K.; Mizukure, T.; Kakizono, D.; Fujihara, K.; Fujii, T.; Mukai, N. New urethanase from the yeast Candida parapsilosis. J. Biosci. Bioeng. 2020, 130, 115–120. [Google Scholar] [CrossRef]
- Lawrence, M.C.; Iliades, P.; Fernley, R.T.; Berglez, J.; Pilling, P.A.; Macreadie, I.G. The Three-dimensional Structure of the Bifunctional 6-Hydroxymethyl-7,8-Dihydropterin Pyrophosphokinase/Dihydropteroate Synthase of Saccharomyces cerevisiae. J. Mol. Biol. 2005, 348, 655–670. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Navarro-Martínez, M.D.; Cabezas-Herrera, J.; Rodríguez-López, J.N. Antifolates as antimycotics? Connection between the folic acid cycle and the ergosterol biosynthesis pathway in Candida albicans. Int. J. Antimicrob. Agents 2006, 28, 560–567. [Google Scholar] [CrossRef]
- Eldesouky, H.E.; Mayhoub, A.; Hazbun, T.R.; Seleem, M. Reversal of Azole Resistance in Candida albicans by Sulfa Antibacterial Drugs. Antimicrob. Agents Chemother. 2018, 62, e00701-17. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Metabolite | g/gDCW | Metabolite | g/gDCW |
|---|---|---|---|
| Lipids | Proteins | ||
| Lanosterol | 0.00063 | L-Valine | 0.03536 |
| Squalene | 0.00017 | L-Tyrosine | 0.02771 |
| Ergosterol | 0.00455 | L-Tryptophan | 0.01356 |
| Phosphatidylserine | 0.00237 | L-Threonine | 0.02230 |
| 1-Phosphatidyl-D-myo-inositol | 0.00173 | L-Serine | 0.02478 |
| Phosphatidylcholine | 0.00288 | L-Proline | 0.01902 |
| Phosphatidylethanolamine | 0.00194 | L-Phenylalanine | 0.02845 |
| Phosphatidic acid | 0.00052 | L-Methionine | 0.04275 |
| Phosphatidylglycerol | 0.00186 | L-Lysine | 0.06440 |
| Tetradecanoic acid | 0.00001 | L-Leucine | 0.03933 |
| Hexadecanoic acid | 0.00074 | L-Isoleucine | 0.02115 |
| (9Z)-Hexadecenoic acid | 0.00010 | L-Histidine | 0.01887 |
| Octadecanoic acid | 0.00032 | L-Glutamate | 0.03987 |
| (9Z)-Octadecenoic acid | 0.00278 | L-Cysteine | 0.00487 |
| (9Z,12Z)-Octadecadienoic acid | 0.00071 | L-Aspartate | 0.00346 |
| (9Z,12Z,15Z)-Octadecatrienoic acid | 0.00016 | L-Asparagine | 0.00362 |
| Triacylglycerol | 0.00467 | L-Arginine | 0.00008 |
| Monoacylglycerol | 0.00401 | L-Alanine | 0.03551 |
| Diacylglycerol | 0.00316 | Glycine | 0.02150 |
| Sterol esters | 0.00445 | L-Glutamine | 0.03987 |
| Soluble Pool | Carbohydrates | ||
| Thiamine | 0.00096 | Chitin | 0.01170 |
| Ubiquinone-6 | 0.00096 | Mannan | 0.23437 |
| NADP+ | 0.00096 | β (1,3)-Glucan | 0.13621 |
| NAD+ | 0.00096 | Deoxyribonucleotides | |
| FMN | 0.00096 | UTP | 0.01599 |
| FAD | 0.00096 | GTP | 0.01378 |
| CoA | 0.00096 | CTP | 0.01313 |
| Biotin | 0.00096 | ATP | 0.01730 |
| Pyridoxal phosphate | 0.00096 | Ribonucleotides | |
| Tetrahydrofolate | 0.00096 | dTTP | 0.00111 |
| dGTP | 0.00074 | ||
| dCTP | 0.00086 | ||
| dATP | 0.00111 | ||
| In Vivo | In Silico | Reference | In Vivo | In Silico | Reference | ||
|---|---|---|---|---|---|---|---|
| Carbon Source | |||||||
| Glucose | + | + | [42,43,44,45,46,47] | L-Sorbose | + | + | [47] |
| Maltose | + | + | [43,47] | D-arabinose | − | − | [43,47] |
| Sucrose | + | + | [42,44,45,46,47] | L-arabinose | + | − | [43,47] |
| Lactose | − | − | [43,45,46,47] | i-Erythritol | − | − | [43,47] |
| Galactose | + | + | [43,47] | Fucose | − | − | [43] |
| Melibiose | − | − | [43,44,46,47] | Salicin | − | − | [43,47] |
| Cellobiose | − | − | [43,47] | Arbutin | − | − | [43,47] |
| Inusitol | − | − | [43,46,47] | D-ribose | + | + | [43,47] |
| Xylose | + | + | [43,45,46,47] | D-Gluconate | + | + | [47] |
| Raffinose | − | − | [43,47] | 2-Keto-D-gluconic acid | + | − | [47] |
| Trehalose | + | + | [43,47] | Inulin | − | − | [47] |
| Galactitol | − | − | [43,45,46,47] | D-Glucosamine | − | − | [47] |
| Rahmnnose | − | − | [43,47] | D-Galacturonate | − | − | [47] |
| Glycerol | + | + | [43,47] | Quinate | − | − | [47] |
| Ribitol | + | − | [43,47] | D-Glucono-1,5-lactone | + | + | [47] |
| Mannitol | + | + | [43,47] | Propane-1,2-diol | − | − | [47] |
| Sorbitol | + | + | [43,47] | D-Glucarate | − | − | [47] |
| Ethanol | + | + | [43,47] | L-Arabinitol | − | − | [47] |
| Methanol | − | − | [47] | D-Glucuronate | − | − | [47] |
| Succinate | + | + | [47] | Butane 2,3 diol | − | − | [47] |
| Lactate | − | − | [47] | D-Galactonate | − | − | [47] |
| Citrate | + | + | [47] | D-Tagaturonate | − | − | [47] |
| Starch | − | − | [47] | Fructose | + | + | [42] |
| Xylitol | + | + | [47] | ||||
| Nitrogen Source | |||||||
| Ammonium | + | + | [47,48] | Urethane | + | + | [49] |
| Citrate | − | − | [47] | Creatine | − | − | [47] |
| L-Lysine | + | + | [47] | Imidazole | − | − | [47] |
| Creatinine | − | − | [47] | L-Glutamate | + | + | [48] |
| D-Tryptophan | − | − | [47] | L-Proline | + | + | [48] |
| Nitrite | − | − | [47] | L-Isoleucine | + | + | [48] |
| Cadaverine | + | + | [47] | Allantoin | + | + | [48] |
| Glucosamine | − | − | [47] | 4-Aminobutanoate | + | + | [48] |
| Ethylamine | + | + | [47] | ||||
| Specific Growth Rate (h−1) | q (mmol g−1 dry weight h−1) | ||||
|---|---|---|---|---|---|
| Glucose | Ethanol | Glycerol | Acetic acid | ||
| In silico | 0.172 | 2.098 | 0 | 0 | 0 |
| In vivo | 0.159 ± 0.027 | 2.098 ± 0.404 | 0 | 0 | 0 |
| Gene Name | EC Number | Gene Name | EC Number | ||||
|---|---|---|---|---|---|---|---|
| C. Parapsilosis | S. Cerevisiae | Human | C. Parapsilosis | S. Cerevisiae | Human | ||
| Homolog | Homolog | Homolog | Homolog | ||||
| CPAR2_302110 | ERG26 | NSDHL | 1.1.1.170 | CPAR2_805350 | PEL1 | PGS1 | 2.7.8.5 |
| CPAR2_104580 | IMD4 | IMPDH | 1.1.1.205 | CPAR2_804250 | PHO8 | ALPL | 3.1.3.1 |
| CPAR2_801560 | ERG27 | DHRS11 | 1.1.1.270 | CPAR2_602700 | GEP4 | PTPMT1 | 3.1.3.27 |
| CPAR2_110330 | HMG1 | HMGCR | 1.1.1.34 | CPAR2_100500 | URA4 | CAD | 3.5.2.3 |
| CPAR2_405900 | ERG24 | TM7SF2 | 1.3.1.70 | CPAR2_806200 | IPP1 | PPA2 | 3.6.1.1 |
| ERG4 | ERG4 | - | 1.3.1.71 | CPAR2_805940 | ADE2 | PAICS | 4.1.1.21 |
| CPAR2_206550 | TMP1 | TYMS | 2.1.1.45 | URA3 | URA3 | UMPS | 4.1.1.23 |
| CPAR2_202250 | ADE17 | ATIC | 2.1.2.3 | CPAR2_109530 | MVD1 | MVD | 4.1.1.33 |
| CPAR2_203160 | URA2 | CAD | 2.1.3.2 | CPAR2_800750 | CAB3 | PPCDC | 4.1.1.36 |
| CPAR2_203160 | URA2 | CAD | 6.3.5.5 | CPAR2_303390 | FOL1 | - | 4.1.2.25 |
| CPAR2_106400 | FKS1 | - | 2.4.1.34 | CPAR2_303390 | FOL1 | - | 2.5.1.15 |
| CPAR2_802790 | URA5 | UMPS | 2.4.2.10 | CPAR2_212310 | ABZ2 | - | 4.1.3.38 |
| CPAR2_208260 | ADE4 | PPAT | 2.4.2.14 | CPAR2_204960 | ADE13 | ADSL | 4.3.2.2 |
| CPAR2_302840 | BTS1 | GGPS1 | 2.5.1.1 | CPAR2_401630 | IDI1 | IDI1 | 5.3.3.2 |
| CPAR2_103950 | ERG20 | FDPS | 2.5.1.10 | CPAR2_301800 | ERG7 | LSS | 5.4.99.7 |
| CPAR2_403110 | ABZ1 | - | 2.6.1.85 | CPAR2_212740 | MET7 | FPGS | 6.3.2.17 |
| CPAR2_502760 | CAB5 | COASY | 2.7.1.24 | CPAR2_500190 | ADE1 | PAICS | 6.3.2.6 |
| CPAR2_202590 | FMN1 | RFK | 2.7.1.26 | CPAR2_208400 | ADE5,7 | GART | 6.3.3.1 |
| CPAR2_602050 | CAB1 | PANK | 2.7.1.33 | CPAR2_208400 | ADE5,7 | GART | 6.3.4.13 |
| CPAR2_105320 | URA6 | CMPK2 | 2.7.4.14 | CPAR2_803640 | ADE12 | ADSS | 6.3.4.4 |
| CPAR2_400710 | ERG8 | PMVK | 2.7.4.2 | CPAR2_204070 | ADE6 | PFAS | 6.3.5.3 |
| CPAR2_304260 | PRS1 | PRPS1 | 2.7.6.1 | CPAR2_804060 | ACC1 | ACACA | 6.4.1.2 |
| CPAR2_500260 | PIS1 | CDIPT | 2.7.8.11 | CPAR2_803530 | ERG12 | MVK | 2.7.1.36 |
| CPAR2_211620 | ADE8 | GART | 2.1.2.2 | CPAR2_701400 | ERG13 | HMGCS | 2.3.3.10 |
| CPAR2_602300 | COQ3 | COQ3 | 2.1.1.114 | FAS1 | FAS1 | - | 2.3.1.86 |
| CPAR2_209250 | COQ5 | COQ5 | 2.1.1.201 | CPAR2_803560 | GUA1 | GMPS | 6.3.5.2 |
| ERG11 | ERG11 | CYP51A1 | 1.14.14.154 | CPAR2_100620 | URA7 | CTPS1 | 6.3.4.2 |
| CPAR2_303080 | GUK1 | GUK1 | 2.7.4.8 | CPAR2_804900 | URA1 | - | 1.3.98.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viana, R.; Couceiro, D.; Carreiro, T.; Dias, O.; Rocha, I.; Teixeira, M.C. A Genome-Scale Metabolic Model for the Human Pathogen Candida Parapsilosis and Early Identification of Putative Novel Antifungal Drug Targets. Genes 2022, 13, 303. https://doi.org/10.3390/genes13020303
Viana R, Couceiro D, Carreiro T, Dias O, Rocha I, Teixeira MC. A Genome-Scale Metabolic Model for the Human Pathogen Candida Parapsilosis and Early Identification of Putative Novel Antifungal Drug Targets. Genes. 2022; 13(2):303. https://doi.org/10.3390/genes13020303
Chicago/Turabian StyleViana, Romeu, Diogo Couceiro, Tiago Carreiro, Oscar Dias, Isabel Rocha, and Miguel Cacho Teixeira. 2022. "A Genome-Scale Metabolic Model for the Human Pathogen Candida Parapsilosis and Early Identification of Putative Novel Antifungal Drug Targets" Genes 13, no. 2: 303. https://doi.org/10.3390/genes13020303
APA StyleViana, R., Couceiro, D., Carreiro, T., Dias, O., Rocha, I., & Teixeira, M. C. (2022). A Genome-Scale Metabolic Model for the Human Pathogen Candida Parapsilosis and Early Identification of Putative Novel Antifungal Drug Targets. Genes, 13(2), 303. https://doi.org/10.3390/genes13020303