
Can the Synergic Contribution of Multigenic Variants Explain the Clinical and Cellular Phenotypes of a Neurodevelopmental Disorder?

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Cell Cultures
2.3. Molecular Studies
2.3.1. Exome Sequencing, Variant Filtering and Prioritization, and CNV Calling
2.3.2. Transcript Analysis in Proband’s Blood and Fibroblasts
2.4. Test for Chromosome Instability Evaluation
2.5. Mitomycin C Sensitility Test
2.6. Complementation with Retroviral Vectors Expressing Functional FANCG
3. Results
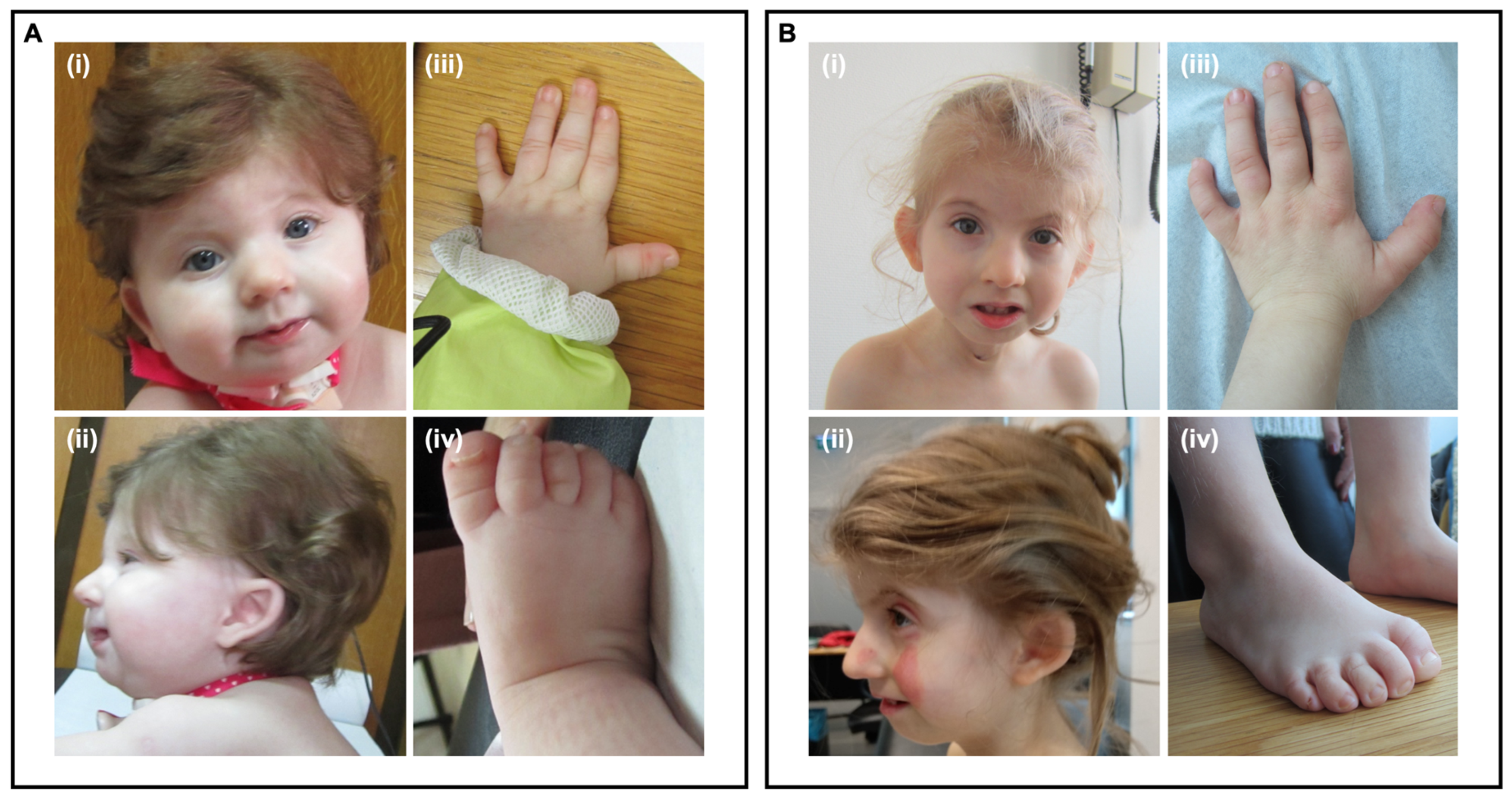
3.1. Clinical Case
3.2. Exome Sequencing Analysis
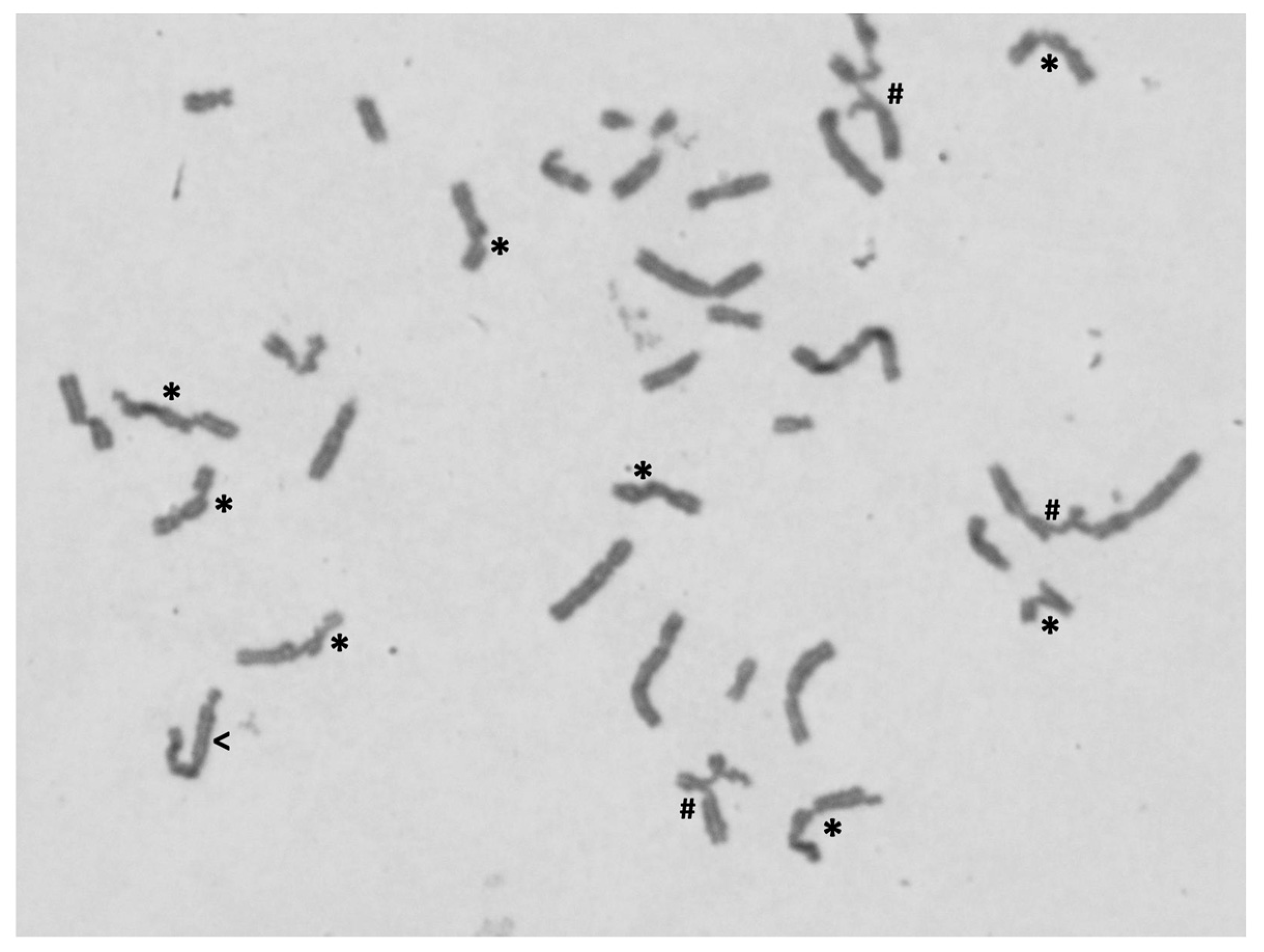
3.3. Chromosome Instability Evaluation
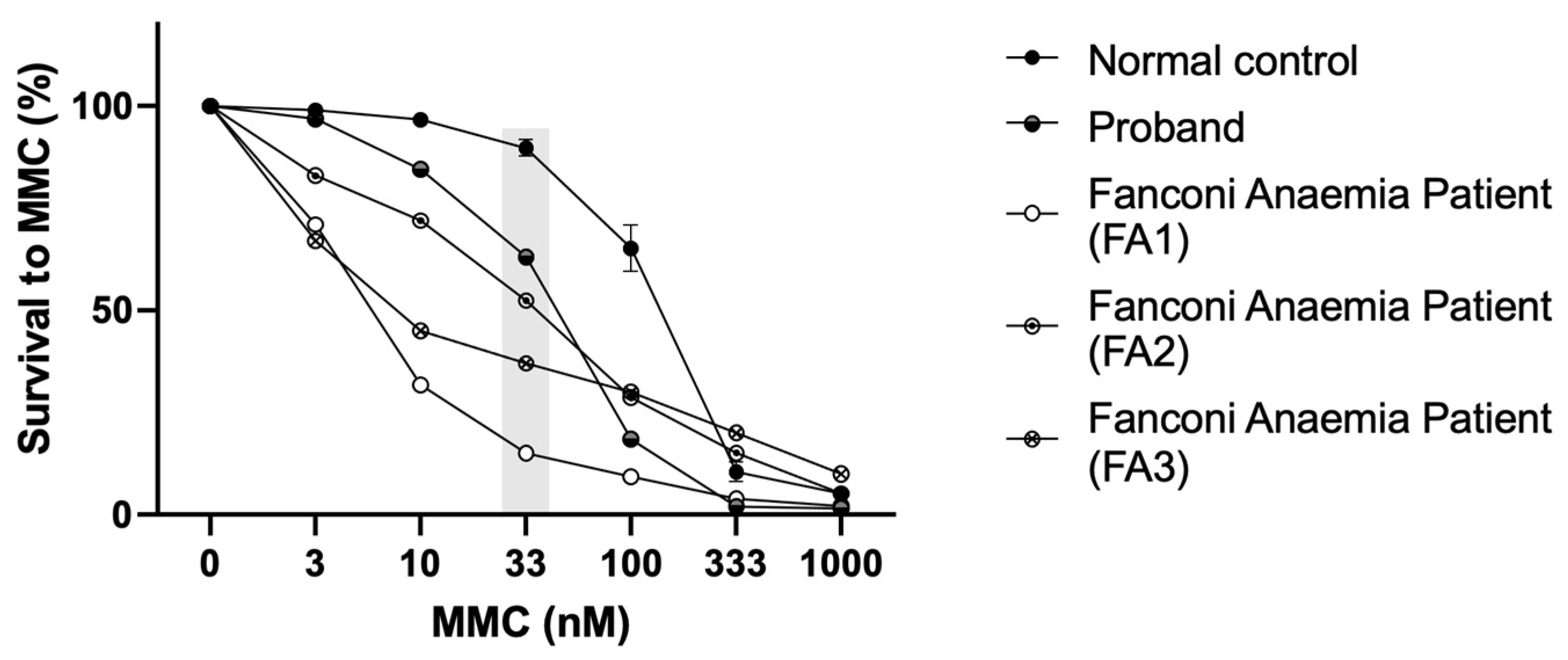
3.4. Mitomycin C Sensitivity Test of Proband’s Cells
3.5. Reanalysis of ES Data Focusing on DNA Repair Genes
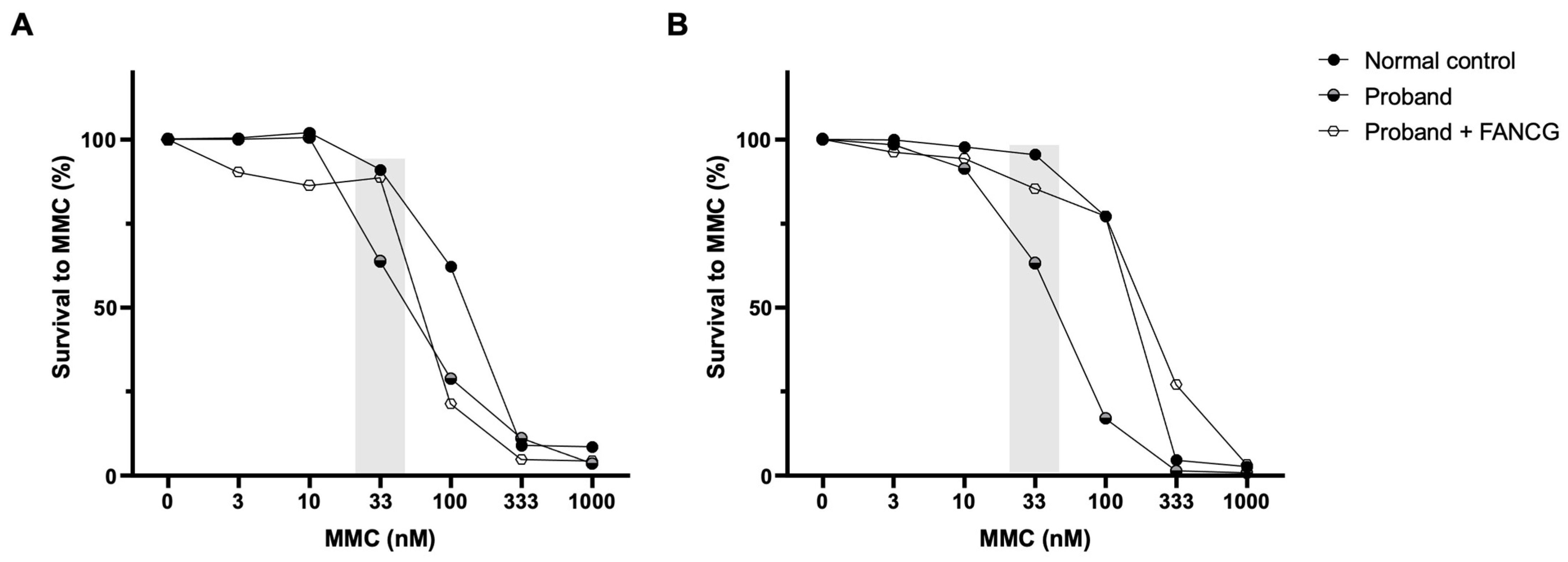
3.6. Complementation Studies with Retroviral Vectors Expressing Functional FANCG
4. Discussion
5. Final Remark
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilfert, A.B.; Sulovari, A.; Turner, T.N.; Coe, B.P.; Eichler, E.E. Recurrent de novo mutations in neurodevelopmental disorders: Properties and clinical implications. Genome Med. 2017, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Association, A.P. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; The American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Parenti, I.; Rabaneda, L.G.; Schoen, H.; Novarino, G. Neurodevelopmental Disorders: From Genetics to Functional Pathways. Trends Neurosci. 2020, 43, 608–621. [Google Scholar] [CrossRef]
- Vissers, L.E.; Gilissen, C.; Veltman, J.A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 2016, 17, 9–18. [Google Scholar] [CrossRef]
- Taniguchi, T.; D’Andrea, A.D. Molecular pathogenesis of Fanconi anemia: Recent progress. Blood 2006, 107, 4223–4233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alter, B.P. Fanconi anemia and the development of leukemia. Best Pr. Res. Clin. Haematol. 2014, 27, 214–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auerbach, A.D. Diagnosis of fanconi anemia by diepoxybutane analysis. Curr. Protoc. Hum. Genet. 2003, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameziane, N.; May, P.; Haitjema, A.; van de Vrugt, H.J.; van Rossum-Fikkert, S.E.; Ristic, D.; Williams, G.J.; Balk, J.; Rockx, D.; Li, H.; et al. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat. Commun. 2015, 6, 8829. [Google Scholar] [CrossRef]
- Gazzo, A.; Raimondi, D.; Daneels, D.; Moreau, Y.; Smits, G.; Van Dooren, S.; Lenaerts, T. Understanding mutational effects in digenic diseases. Nucleic Acids Res. 2017, 45, e140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadimitriou, S.; Gazzo, A.; Versbraegen, N.; Nachtegael, C.; Aerts, J.; Moreau, Y.; Van Dooren, S.; Nowe, A.; Smits, G.; Lenaerts, T. Predicting disease-causing variant combinations. Proc. Natl. Acad. Sci. USA 2019, 116, 11878–11887. [Google Scholar] [CrossRef] [Green Version]
- Gazzo, A.M.; Daneels, D.; Cilia, E.; Bonduelle, M.; Abramowicz, M.; Van Dooren, S.; Smits, G.; Lenaerts, T. DIDA: A curated and annotated digenic diseases database. Nucleic Acids Res. 2016, 44, D900–D907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, M.B.; Senapathy, P. RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987, 15, 7155–7174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Lin, X.; Salzberg, S.L. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 2001, 29, 1185–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Dunnen, J.T.; Antonarakis, S.E. Nomenclature for the description of human sequence variations. Hum. Genet. 2001, 109, 121–124. [Google Scholar] [CrossRef] [PubMed]
- den Dunnen, J.T.; Antonarakis, S.E. Mutation nomenclature extensions and suggestions to describe complex mutations: A discussion. Hum. Mutat. 2000, 15, 7–12. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Krumm, N.; Sudmant, P.H.; Ko, A.; O’Roak, B.J.; Malig, M.; Coe, B.P.; Quinlan, A.R.; Nickerson, D.A.; Eichler, E.E. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012, 22, 1525–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auerbach, A.D.; Rogatko, A.; Schroeder-Kurth, T.M. International Fanconi Anemia Registry: Relation of clinical symptoms to diepoxybutane sensitivity. Blood 1989, 73, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Antonio Casado, J.; Callén, E.; Jacome, A.; Río, P.; Castella, M.; Lobitz, S.; Ferro, T.; Muñoz, A.; Sevilla, J.; Cantalejo, A.; et al. A comprehensive strategy for the subtyping of patients with Fanconi anaemia: Conclusions from the Spanish Fanconi Anemia Research Network. J. Med. Genet. 2007, 44, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Hanenberg, H.; Batish, S.D.; Pollok, K.E.; Vieten, L.; Verlander, P.C.; Leurs, C.; Cooper, R.J.; Gottsche, K.; Haneline, L.; Clapp, D.W.; et al. Phenotypic correction of primary Fanconi anemia T cells with retroviral vectors as a diagnostic tool. Exp. Hematol. 2002, 30, 410–420. [Google Scholar] [CrossRef]
- Vilarinho, L.; Rocha, H.; Sousa, C.; Marcao, A.; Fonseca, H.; Bogas, M.; Osorio, R.V. Four years of expanded newborn screening in Portugal with tandem mass spectrometry. J. Inherit. Metab. Dis. 2010, 33 (Suppl. 3), S133–S138. [Google Scholar] [CrossRef]
- Harakalova, M.; van den Boogaard, M.J.; Sinke, R.; van Lieshout, S.; van Tuil, M.C.; Duran, K.; Renkens, I.; Terhal, P.A.; de Kovel, C.; Nijman, I.J.; et al. X-exome sequencing identifies a HDAC8 variant in a large pedigree with X-linked intellectual disability, truncal obesity, gynaecomastia, hypogonadism and unusual face. J. Med. Genet. 2012, 49, 539–543. [Google Scholar] [CrossRef]
- Kaiser, F.J.; Ansari, M.; Braunholz, D.; Concepcion Gil-Rodriguez, M.; Decroos, C.; Wilde, J.J.; Fincher, C.T.; Kaur, M.; Bando, M.; Amor, D.J.; et al. Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Hum. Mol. Genet. 2014, 23, 2888–2900. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Zhou, D.; Zhang, Z.; Liu, Y.; Yang, Y. Exome sequencing identifies a de novo mutation in HDAC8 associated with Cornelia de Lange syndrome. J. Hum. Genet. 2014, 59, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Fieremans, N.; Van Esch, H.; Holvoet, M.; Van Goethem, G.; Devriendt, K.; Rosello, M.; Mayo, S.; Martinez, F.; Jhangiani, S.; Muzny, D.M.; et al. Identification of Intellectual Disability Genes in Female Patients with A Skewed X Inactivation Pattern. Hum. Mutat. 2016, 37, 804–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parenti, I.; Gervasini, C.; Pozojevic, J.; Wendt, K.S.; Watrin, E.; Azzollini, J.; Braunholz, D.; Buiting, K.; Cereda, A.; Engels, H.; et al. Expanding the clinical spectrum of the ‘HDAC8-phenotype’—Implications for molecular diagnostics, counseling and risk prediction. Clin. Genet. 2016, 89, 564–573. [Google Scholar] [CrossRef]
- Gao, X.; Huang, Z.; Fan, Y.; Sun, Y.; Liu, H.; Wang, L.; Gu, X.F.; Yu, Y. A Functional Mutation in HDAC8 Gene as Novel Diagnostic Marker for Cornelia De Lange Syndrome. Cell. Physiol. Biochem. 2018, 47, 2388–2395. [Google Scholar] [CrossRef]
- Vannini, A.; Volpari, C.; Filocamo, G.; Casavola, E.C.; Brunetti, M.; Renzoni, D.; Chakravarty, P.; Paolini, C.; De Francesco, R.; Gallinari, P.; et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 15064–15069. [Google Scholar] [CrossRef] [Green Version]
- Migeon, B.R. X-linked diseases: Susceptible females. Genet. Med. Off. J. Am. Coll. Med. Genet. 2020, 22, 1156–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, R.; Buchwald, M. Susceptibility of Fanconi’s anemia lymphoblasts to DNA-cross-linking and alkylating agents. Cancer Res. 1982, 42, 4000–4006. [Google Scholar]
- Frohnmayer, L.V.R.S.; Wirkkula, L. Fanconi Anemia Clinical Care Guidelines, 5th ed.; Fanconi Anemia Research Fund: Eugene, OR, USA, 2020. [Google Scholar]
- Belo, H.; Silva, G.; Cardoso, B.A.; Porto, B.; Minguillon, J.; Barbot, J.; Coutinho, J.; Casado, J.A.; Benedito, M.; Saturnino, H.; et al. Epigenetic Alterations in Fanconi Anaemia: Role in Pathophysiology and Therapeutic Potential. PLoS ONE 2015, 10, e0139740. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Group | Mean | Min | Max |
|---|---|---|---|---|
| Nr breaks/cell | FA | 8.96 | 1.30 | 23.90 |
| Non-FA/control | 0.06 | 0.00 | 0.36 | |
| % ab cells | FA | 85.15 | 12.60 | 100.00 |
| Non-FA/control | 5.12 | 0.00 | 22.00 | |
| Difference between FA and non-FA/control groups for the two parameters: number of breaks per cell (Nr breaks/cell) and percentage of aberrant cell, i.e., cells with breaks (% ab cells). | ||||
| DEB-Induced CI | DEB Concentration: 0.05 µg/mL (Diagnostic Discriminative Parameters to Compare with Reference Values in Table 1) | DEB Concentration 0.1 µg/mL | ||
|---|---|---|---|---|
| % ab Cells | Nr Breaks/Cell | % ab Cells | Nr Breaks/Cell | |
| Proband | 34 | 0.54 | 76 | 1.88 |
| Mother’s Proband | 4 | 0.06 | 5 | 0.05 |
| Father’s Proband | 1 | 0.01 | 3 | 0.03 |
| Healthy Donor (Negative Control) | 3 | 0.04 | 2 | 0.02 |
| FA Patient (Positive Control) | 89 | 5.36 | No Metaphases | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maia, N.; Nabais Sá, M.J.; Oliveira, C.; Santos, F.; Soares, C.A.; Prior, C.; Tkachenko, N.; Santos, R.; de Brouwer, A.P.M.; Jacome, A.; et al. Can the Synergic Contribution of Multigenic Variants Explain the Clinical and Cellular Phenotypes of a Neurodevelopmental Disorder? Genes 2022, 13, 78. https://doi.org/10.3390/genes13010078
Maia N, Nabais Sá MJ, Oliveira C, Santos F, Soares CA, Prior C, Tkachenko N, Santos R, de Brouwer APM, Jacome A, et al. Can the Synergic Contribution of Multigenic Variants Explain the Clinical and Cellular Phenotypes of a Neurodevelopmental Disorder? Genes. 2022; 13(1):78. https://doi.org/10.3390/genes13010078
Chicago/Turabian StyleMaia, Nuno, Maria João Nabais Sá, Cláudia Oliveira, Flávia Santos, Célia Azevedo Soares, Catarina Prior, Nataliya Tkachenko, Rosário Santos, Arjan P. M. de Brouwer, Ariana Jacome, and et al. 2022. "Can the Synergic Contribution of Multigenic Variants Explain the Clinical and Cellular Phenotypes of a Neurodevelopmental Disorder?" Genes 13, no. 1: 78. https://doi.org/10.3390/genes13010078
APA StyleMaia, N., Nabais Sá, M. J., Oliveira, C., Santos, F., Soares, C. A., Prior, C., Tkachenko, N., Santos, R., de Brouwer, A. P. M., Jacome, A., Porto, B., & Jorge, P. (2022). Can the Synergic Contribution of Multigenic Variants Explain the Clinical and Cellular Phenotypes of a Neurodevelopmental Disorder? Genes, 13(1), 78. https://doi.org/10.3390/genes13010078