
Review of the Forensic Applicability of Biostatistical Methods for Inferring Ancestry from Autosomal Genetic Markers


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Ancestry Informative Markers
2. Biostatistical Methods
2.1. Principal Components Analysis
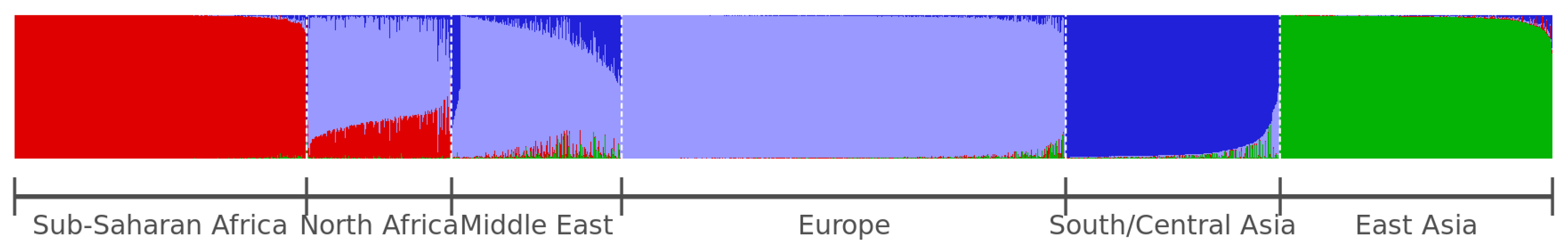
2.2. Model-Based Clustering Methods
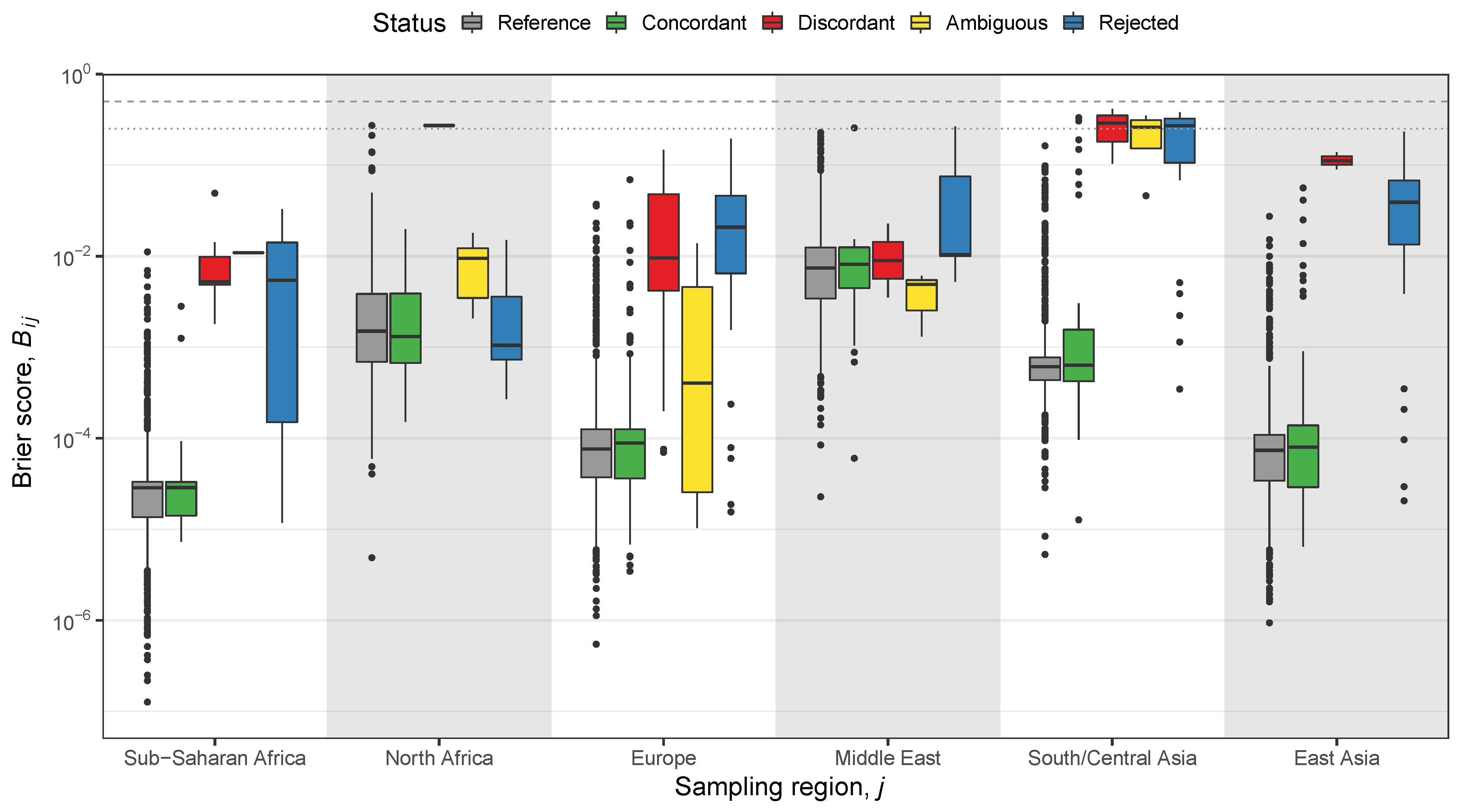
2.3. Classification and Likelihood-Based Methods
2.4. Hypothesis Test-Based Methods
3. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International HapMap Consortium. The International HapMap Project. Nature 2003, 426, 789–796. [Google Scholar] [CrossRef] [PubMed]
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Mallick, S.; Li, H.; Lipson, M.; Mathieson, I.; Gymrek, M.; Racimo, F.; Zhao, M.; Chennagiri, N.; Nordenfelt, S.; Tandon, A.; et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 2016, 538, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Cavalli-Sforza, L.L.; Menozzi, P.; Piazza, A. The History and Geography of Human Genes; Princeton Universily Press: Princeton, NJ, USA, 1994. [Google Scholar]
- Jobling, M.A.; Hollox, E.; Hurles, M.; Kivisild, T.; Tyler-Smith, C. Human Evolutionary Genetics, 2nd ed.; Garland Science Taylor & Francis Group: New York, NY, USA, 2014. [Google Scholar]
- Rosenberg, N.A.; Pritchard, J.K.; Weber, J.L.; Cann, H.M.; Kidd, K.K.; Zhivotovsky, L.A.; Feldman, M.W. Genetic structure of human populations. Science 2002, 298, 2381–2384. [Google Scholar] [CrossRef] [PubMed]
- Cavalli-Sforza, L.L.; Feldman, M.W. The application of molecular genetic approaches to the study of human evolution. Nat. Genet. (Suppl.) 2003, 33, 266–275. [Google Scholar] [CrossRef]
- Serre, D.; Pääbo, S. Evidence for gradients of human genetic diversity within and among continents. Genome Res. 2004, 14, 1679–1685. [Google Scholar] [CrossRef]
- Manica, A.; Prugnolle, F.; Balloux, F. Geography is a better determinant of genetic differentiation than ethnicity. Hum. Genet. 2005, 118, 366–371. [Google Scholar] [CrossRef]
- Novembre, J.; Johnson, T.; Bryc, K.; Kutalik, Z.; Boyko, A.R.; Auton, A.; Indap, A.; King, K.S.; Bergmann, S.; Nelson, M.R.; et al. Genes mirror geography within Europe. Nature 2008, 456, 98–101. [Google Scholar] [CrossRef]
- Wang, C.; Zöllner, S.; Rosenberg, N. A quantitative comparison of the similarity between genes and geography in worldwide human populations. PLoS Genet. 2012, 8, e1002886. [Google Scholar] [CrossRef]
- Rosenberg, N.; Li, L.; Ward, R.; Pritchard, J. Informativeness of genetic markers for inference of ancestry. Am. J. Hum. Genet. 2003, 73, 1402–1422. [Google Scholar] [CrossRef]
- Rosenberg, N.A. Algorithms for Selecting Informative Marker Panels for Population Assignment. J. Comput. Biol. 2005, 12, 1183–1201. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, B.; Junge, A.; Meyer, E.; Wiegand, P. Population Genetic Diversity in Relation to Microsatellite Heterogeneity. Hum. Mut. 1998, 11, 135–144. [Google Scholar] [CrossRef]
- Alladio, E.; Della Rocca, C.; Barni, F.; Dugoujon, J.M.; Garofano, P.; Semino, O.; Berti, A.; Novelletto, A.; Vincenti, M.; Cruciani, F.; et al. A multivariate statistical approach for the estimation of the ethnic origin of unknown genetic profiles in forensic genetics. Forensic Sci. Int. Genet. 2020, 45, 102209. [Google Scholar] [CrossRef]
- Oldoni, F.; Kidd, K.K.; Podini, D. Microhaplotypes in forensic genetics. Forensic Sci. Int. Genet. 2019, 38, 54–69. [Google Scholar] [CrossRef]
- Yang, N.; Li, H.; Criswell, L.A.; Gregersen, P.K.; Alarcon-Riquelme, M.E.; Kittles, R.; Shigeta, R.; Silva, G.; Patel, P.I.; Belmont, J.W.; et al. Examination of ancestry and ethnic affiliation using highly informative diallelic DNA markers: Application to diverse and admixed populations and implications for clinical epidemiology and forensic medicine. Hum. Genet. 2005, 118, 382–392. [Google Scholar] [CrossRef]
- Moriot, A.; Santos, C.; Freire-Aradas, A.; Phillips, C.; Hall, D. Inferring biogeographic ancestry with compound markers of slow and fast evolving polymorphisms. Eur. J. Hum. Genet. 2018, 26, 1697–1707. [Google Scholar] [CrossRef]
- Phillips, C. Forensic genetic analysis of bio-geographical ancestry. Forensic Sci. Int. Genet. 2015, 18, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.; Santos, C.; Fondevila, M.; Carracedo, Á.; Lareu, M.V. Inference of Ancestry in Forensic Analysis I: Autosomal Ancestry-Informative Marker Sets. In Forensic DNA Typing Protocols; Methods in Molecular Biology; Goodwin, W., Ed.; Springer: Berlin/Heidelberg, Germany, 2016; Volume 1420, pp. 234–253. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of Population Structure Using Multilocus Genotype Data: Linked Loci and Correlated Allele Frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef]
- Lindskou, M.; Eriksen, P.S.; Tvedebrink, T. Outlier detection in contingency tables using decomposable graphical models. Scand. J. Stat. 2020, 47, 347–360. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Patterson, N.; Price, A.L.; Reich, D. Population Structure and Eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- Mogensen, H.S.; Tvedebrink, T.; Børsting, C.; Pereira, V.; Morling, N. Ancestry prediction efficiency of the software GenoGeographer using a z-score method and the ancestry informative markers in the Precision ID Ancestry Panel. Forensic Sci. Int. Genet. 2020, 44, 102154. [Google Scholar] [CrossRef]
- Menozzi, P.; Piazza, A.; Cavalli-Sforza, L.L. Synthetic maps of human gene frequencies in Europeans. Science 1978, 201, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Novembre, J.; Stephens, M. Interpreting principal component analyses of spatial population genetic variation. Nat. Genet. 2008, 40, 646–649. [Google Scholar] [CrossRef]
- McVean, G. A Genealogical Interpretation of Principal Components Analysis. PLoS Genet. 2009, 5, 1–10. [Google Scholar] [CrossRef]
- Wangkumhang, P.; Hellenthal, G. Statistical methods for detecting admixture. Curr. Opin. Genet. Dev. 2018, 53, 121–127. [Google Scholar] [CrossRef]
- Miller, J.M.; Cullingham, C.I.; Peery, R.M. The influence of a priori grouping on inference of genetic clusters: Simulation study and literature review of the DAPC method. Heredity 2020, 125, 269–280. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P.J. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Novembre, J. Pritchard, Stephens, and Donnelly on Population Structure. Genetics 2016, 204, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Rannala, B.; Mountain, J.L. Detecting immigration by using multilocus genotypes. Proc. Natl. Acad. Sci. USA 1997, 94, 9197–9201. [Google Scholar] [CrossRef] [PubMed]
- Foreman, L.; Smith, A.; Evett, I. Bayesian analysis of DNA profiling data in forensic identification applications. J. R. Stat. Soc. A 1997, 160, 429–469. [Google Scholar] [CrossRef]
- Roeder, K.; Escobar, M.; Kadane, J.B.; Balazs, I. Measuring heterogeneity in forensic databases using hierarchical Bayes models. Biometrika 1998, 85, 269–287. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef]
- Tang, H.; Peng, J.; Wang, P.; Risch, N. Estimation of Individual Admixture: Analytical and Study Design Considerations. Genet. Epidemiol. 2005, 28, 289–301. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Raj, A.; Stephens, M.; Pritchard, J.K. fastSTRUCTURE: Variational Inference of Population Structure in Large SNP Data Sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef]
- Novembre, J. Variations on a Common STRUCTURE: New Algorithms for a Valuable Model. Genetics 2014, 197, 809–811. [Google Scholar] [CrossRef]
- Lawson, D.J.; van Dorp, L.; Falush, D. A tutorial on how not to over-interpret STRUCTURE and ADMIXTURE bar plots. Nat. Commun. 2018, 9, 3258. [Google Scholar] [CrossRef]
- Lawson, D. badMIXTURE: Validating Structure With Chromosome Painting; R Package Version 0.0.0.9000. 2018. Available online: https://github.com/danjlawson/badMIXTURE (accessed on 13 December 2021).
- Hellenthal, G.; Busby, G.B.J.; Band, G.; Wilson, J.F.; Capelli, C.; Falush, D.; Myers, S. A Genetic Atlas of Human Admixture History. Science 2014, 343, 747–751. [Google Scholar] [CrossRef]
- Lawson, D.J.; Hellenthal, G.; Myers, S.; Falush, D. Inference of Population Structure using Dense Haplotype Data. PLoS Genet. 2012, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef]
- Cheung, K.H.; Osier, M.V.; Kidd, J.R.; Pakstis, A.J.; Miller, P.L.; Kidd, K.K. ALFRED: An allele frequency database for diverse populations and DNA polymorphisms. Nucleic Acids Res. 2000, 28, 361–363. [Google Scholar] [CrossRef][Green Version]
- Pakstis, A.; Kang, L.; Liu, L.; Zhang, Z.; Jin, T.; Grigorenko, E.L.; Wendt, F.R.; Budowle, B.; Hadi, S.; Qahtanif, M.S.A.; et al. Increasing the reference populations for the 55 AISNP panel: The need and benefits. Int. J. Legal Med. 2017, 131, 913–917. [Google Scholar] [CrossRef]
- Phillips, C.; Prieto, L.; Fondevila, M.; Salas, A.; Gomez-Tato, A.; Alvarez-Dios, J.; Alonso, A.; Blanco-Verea, A.; Brion, M.; Montesino, M.; et al. Ancestry analysis in the 11-M Madrid bomb attack investigation. PLoS ONE 2009, 4, e6583. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.Y.; Gahan, M.E.; McNevin, D. Prediction of biogeographical ancestry from genotype: A comparison of classifiers. Int. J. Legal Med. 2017, 131, 901–912. [Google Scholar] [CrossRef]
- McNevina, D.; Santos, C.; Gómez-Tato, A.; Álvarez-Dios, J.; Casares de Cal, M.; Daniel, R.; Phillips, C.; Lareu, M.V. Anassessment of Bayesian and multinomial logistic regression classification systems to analyse admixed individuals. Forensic Sci. Int. Genet. Suppl. Ser. 2013, 4, e63–e64. [Google Scholar] [CrossRef]
- Cheung, E.Y.Y.; Gahan, M.E.; McNevin, D. Prediction of biogeographical ancestry in admixed individuals. Forensic Sci. Int. Genet. 2018, 36, 104–111. [Google Scholar] [CrossRef]
- Tvedebrink, T.; Eriksen, P.S.; Mogensen, H.S.; Morling, N. Weight of the Evidence of Genetic Investigations of Ancestry Informative Markers. Theor. Popul. Biol. 2018, 120, 1–10. [Google Scholar] [CrossRef]
- Tvedebrink, T.; Eriksen, P.S. Inference of admixed ancestry with Ancestry Informative Markers. Forensic Sci. Int. Genet. 2019, 42, 147–153. [Google Scholar] [CrossRef]
- Pfaffelhuber, P.; Sester-Huss, E.; Baumdicker, F.; Naue, J.; Lutz-Bonengel, S.; Staubach, F. Inference of recent admixture using genotype data. Forensic Sci. Int. Genet. 2021, 56, 102593. [Google Scholar] [CrossRef]
- Tvedebrink, T.; Eriksen, P.S.; Mogensen, H.S.; Morling, N. GenoGeographer—A tool for genogeographic inference. Forensic Sci. Int. Genet. Suppl. Ser. 2017, 6, e463–e465. [Google Scholar] [CrossRef]
- Kling, D.; Tillmar, A.; Egeland, T.; Mostad, P. A general model for likelihood computations of genetic marker data accounting for linkage, linkage disequilibrium, and mutations. Int. J. Legal Med. 2015, 129, 943–954. [Google Scholar] [CrossRef]
- Porras-Hurtado, L.; Ruiz, Y.; Santos, C.; Phillips, C.; Carracedo, Á.; Lareu, M. An overview of STRUCTURE: Applications, parameter settings, and supporting software. Front. Genet. 2013, 4, 98. [Google Scholar] [CrossRef]
- Santos, C.; Phillips, C.; Gomez-Tato, A.; Alvarez-Dios, J.; Carracedo, Á.; Lareu, M.V. Inference of Ancestry in Forensic Analysis II: Analysis of Genetic Data. In Forensic DNA Typing Protocols; Methods in Molecular Biology; Goodwin, W., Ed.; Springer: Berlin/Heidelberg, Germany, 2016; Volume 1420, pp. 255–285. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tvedebrink, T. Review of the Forensic Applicability of Biostatistical Methods for Inferring Ancestry from Autosomal Genetic Markers. Genes 2022, 13, 141. https://doi.org/10.3390/genes13010141
Tvedebrink T. Review of the Forensic Applicability of Biostatistical Methods for Inferring Ancestry from Autosomal Genetic Markers. Genes. 2022; 13(1):141. https://doi.org/10.3390/genes13010141
Chicago/Turabian StyleTvedebrink, Torben. 2022. "Review of the Forensic Applicability of Biostatistical Methods for Inferring Ancestry from Autosomal Genetic Markers" Genes 13, no. 1: 141. https://doi.org/10.3390/genes13010141
APA StyleTvedebrink, T. (2022). Review of the Forensic Applicability of Biostatistical Methods for Inferring Ancestry from Autosomal Genetic Markers. Genes, 13(1), 141. https://doi.org/10.3390/genes13010141