
Genetic Regulation of Cytokine Response in Patients with Acute Community-Acquired Pneumonia

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Sample
2.2. Cytokine Measurement and Analysis
2.3. Genotyping, Quality Control, and Imputation
2.4. Genome-Wide Association Study
2.5. Conditional and Joint Analysis
2.6. Credible Set Analysis
2.7. Colocalisation Analysis
2.8. MetaXcan Analysis
2.9. Lookup of Cytokine Coding Genes
3. Results
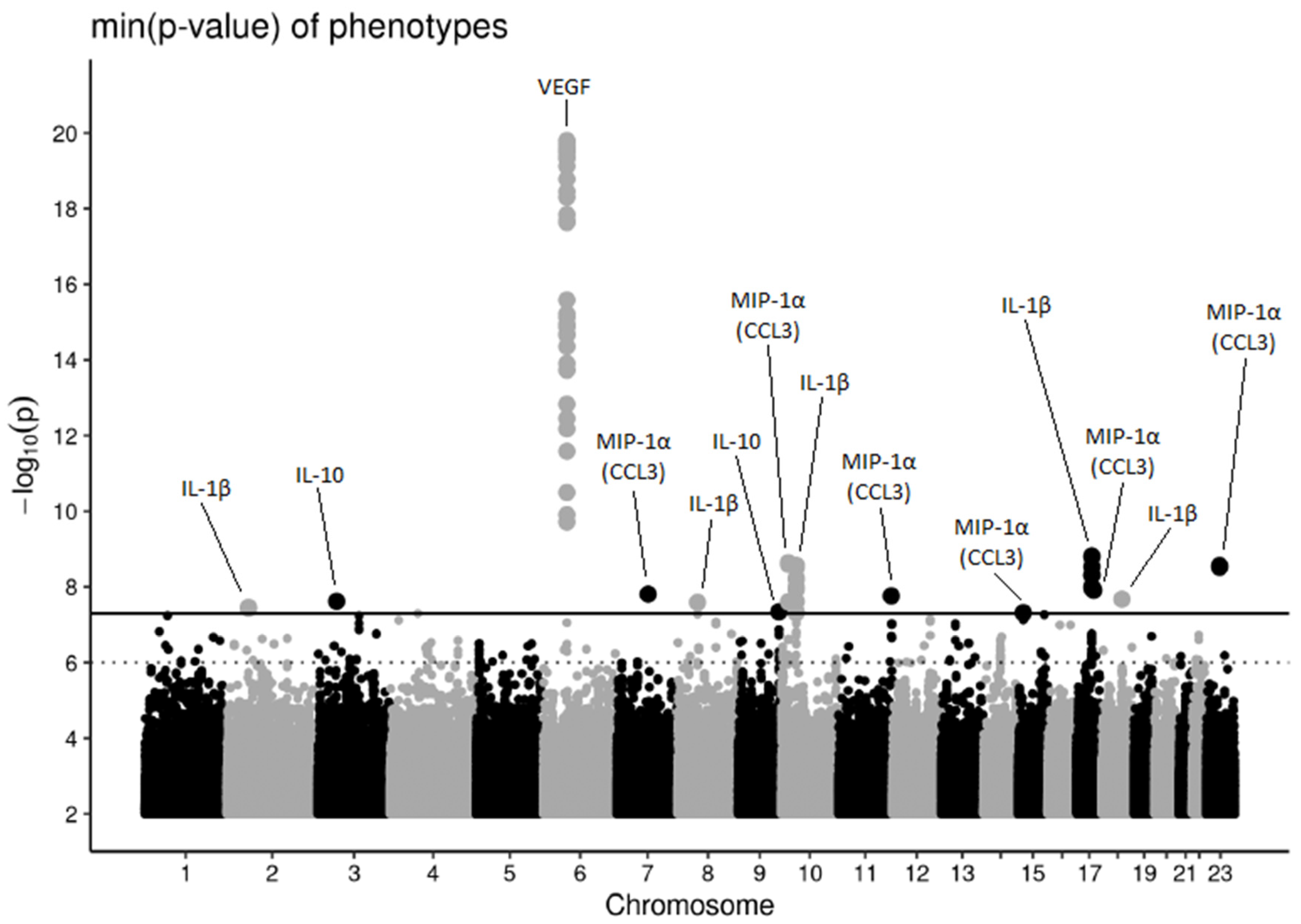
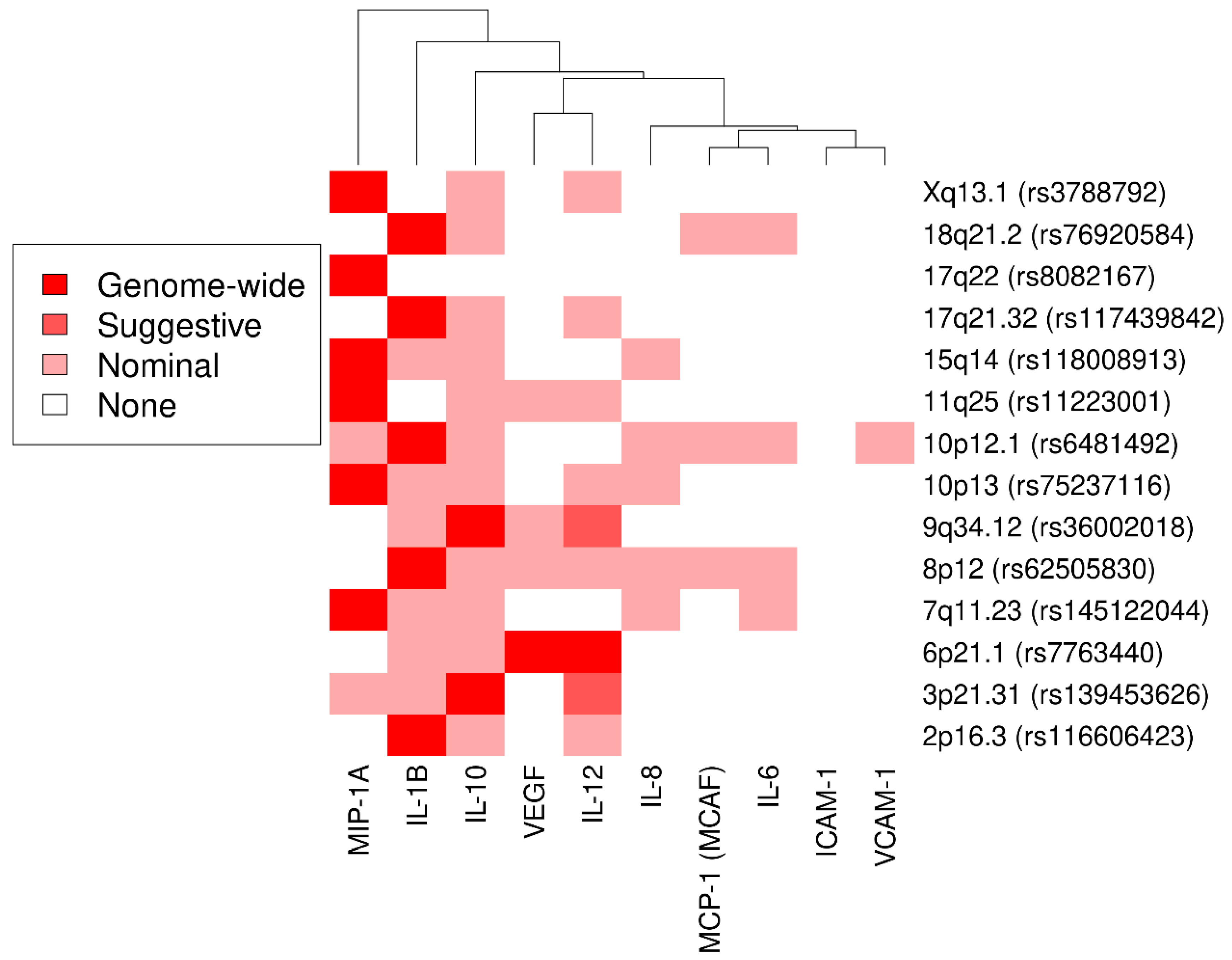
3.1. Genome-Wide Association Study and Secondary Analyses
3.2. Known Associations
3.2.1. Vascular Endothelial Growth Factor and Interleukin 12
3.2.2. Interleukin 1β
3.3. Novel Associations
3.3.1. Interleukin 1β
3.3.2. Interleukin 10
3.3.3. Macrophage Inflammatory Protein 1α
3.4. Lookup of Cytokine-Coding Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lanks, C.W.; Musani, A.I.; Hsia, D.W. Community-acquired Pneumonia and Hospital-acquired Pneumonia. Med. Clin. N. Am. 2019, 103, 487–501. [Google Scholar] [CrossRef]
- Curran, P.J.; Howard, A.L.; Bainter, S.A.; Lane, S.T.; McGinley, J.S. The separation of between-person and within-person components of individual change over time: A latent curve model with structured residuals. J. Consult. Clin. Psychol. 2014, 82, 879–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioanas, M.; Ferrer, M.; Cavalcanti, M.; Ferrer, R.; Ewig, S.; Filella, X.; de La Bellacasa, J.P.; Torres, A. Causes and predictors of nonresponse to treatment of intensive care unit-acquired pneumonia. Crit. Care Med. 2004, 32, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Serrano, S.; Dorca, J.; Coromines, M.; Carratalà, J.; Gudiol, F.; Manresa, F. Molecular inflammatory responses measured in blood of patients with severe community-acquired pneumonia. Clin. Vaccine Immunol. 2003, 10, 813–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosolowski, M.; Oberle, V.; Ahnert, P.; Creutz, P.; Witzenrath, M.; Kiehntopf, M.; Loeffler, M.; Suttorp, N.; Scholz, M. Dynamics of cytokines, immune cell counts and disease severity in patients with community-acquired pneumonia—Unravelling potential causal relationships. Cytokine 2020, 136, 155263. [Google Scholar] [CrossRef] [PubMed]
- Georgakis, M.K.; Gill, D.; Rannikmäe, K.; Traylor, M.; Anderson, C.D.; Lee, J.-M.; Kamatani, Y.; Hopewell, J.C.; Worrall, B.B.; Bernhagen, J.; et al. Genetically Determined Levels of Circulating Cytokines and Risk of Stroke. Circulation 2019, 139, 256–268. [Google Scholar] [CrossRef]
- Bek, S.; Bojesen, A.B.; Nielsen, J.V.; Sode, J.; Bank, S.; Vogel, U.; Andersen, V. Systematic review and meta-analysis: Pharmacogenetics of anti-TNF treatment response in rheumatoid arthritis. Pharm. J. 2017, 17, 403–411. [Google Scholar] [CrossRef]
- Ahola-Olli, A.V.; Würtz, P.; Havulinna, A.S.; Aalto, K.; Pitkänen, N.; Lehtimäki, T.; Kähönen, M.; Lyytikäinen, L.-P.; Raitoharju, E.; Seppälä, I.; et al. Genome-wide Association Study Identifies 27 Loci Influencing Concentrations of Circulating Cytokines and Growth Factors. Am. J. Hum. Genet. 2017, 100, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, L.; Yang, Y.-H.; He, L.; Chou, K.-C. Modulation of Cytokine Network in the Comorbidity of Schizophrenia and Tuberculosis. Curr. Top. Med. Chem. 2016, 16, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.P.; Ritchie, S.C.; Grinberg, N.F.; Tang, H.H.-F.; Huang, Q.Q.; Teo, S.M.; Ahola-Olli, A.V.; Würtz, P.; Havulinna, A.S.; Santalahti, K.; et al. Multivariate Genome-wide Association Analysis of a Cytokine Network Reveals Variants with Widespread Immune, Haematological, and Cardiometabolic Pleiotropy. Am. J. Hum. Genet. 2019, 105, 1076–1090. [Google Scholar] [CrossRef] [PubMed]
- Ahnert, P.; Creutz, P.; Scholz, M.; Schütte, H.; Engel, C.; Hossain, H.; Chakraborty, T.; Bauer, M.; Kiehntopf, M.; Völker, U.; et al. PROGRESS—Prospective observational study on hospitalized community acquired pneumonia. BMC Pulm. Med. 2016, 16, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahnert, P.; Creutz, P.; Horn, K.; Schwarzenberger, F.; Kiehntopf, M.; Hossain, H.; Bauer, M.; Brunkhorst, F.M.; Reinhart, K.; Völker, U.; et al. Sequential organ failure assessment score is an excellent operationalization of disease severity of adult patients with hospitalized community acquired pneumonia—Results from the prospective observational PROGRESS study. Crit. Care 2019, 23, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aken, B.L.; Achuthan, P.; Akanni, W.; Amode, M.R.; Bernsdorff, F.; Bhai, J.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; et al. Ensembl 2017. Nucleic Acids Res. 2017, 45, D635–D642. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J.; et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017, 45, D896–D901. [Google Scholar] [CrossRef] [PubMed]
- Human Genomics. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirsten, H.; Al-Hasani, H.; Holdt, L.; Gross, A.; Beutner, F.; Krohn, K.; Horn, K.; Ahnert, P.; Burkhardt, R.; Reiche, K.; et al. Dissecting the genetics of the human transcriptome identifies novel trait-related trans-eQTLs and corroborates the regulatory relevance of non-protein coding loci†. Hum. Mol. Genet. 2015, 24, 4746–4763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakefield, J. Bayes factors for genome-wide association studies: Comparison with P-values. Genet. Epidemiol. 2009, 33, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, J. A Bayesian measure of the probability of false discovery in genetic epidemiology studies. Am. J. Hum. Genet. 2007, 81, 208–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GTEx. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Barbeira, A.N.; Dickinson, S.P.; Bonazzola, R.; Zheng, J.; Wheeler, H.E.; Torres, J.M.; Torstenson, E.S.; Shah, K.P.; Garcia, T.; Edwards, T.L.; et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 2018, 9, 1825. [Google Scholar] [CrossRef]
- Maffioletti, E.; Gennarelli, M.; Magri, C.; Bocchio-Chiavetto, L.; Bortolomasi, M.; Bonvicini, C.; Abate, M.; Trabucchi, L.; Ulivi, S.; Minelli, A. Genetic determinants of circulating VEGF levels in major depressive disorder and electroconvulsive therapy response. Drug Dev. Res. 2020, 81, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Kechris, K.; Jacobson, S.; Drummond, M.B.; Hawkins, G.A.; Yang, J.; Chen, T.; Quibrera, P.M.; Anderson, W.; Barr, R.G.; et al. Common Genetic Polymorphisms Influence Blood Biomarker Measurements in COPD. PLoS Genet. 2016, 12, e1006011. [Google Scholar] [CrossRef]
- Jin, G.; Ma, H.; Wu, C.; Dai, J.; Zhang, R.; Shi, Y.; Lu, J.; Miao, X.; Wang, M.; Zhou, Y.; et al. Genetic variants at 6p21.1 and 7p15.3 are associated with risk of multiple cancers in Han Chinese. Am. J. Hum. Genet. 2012, 91, 928–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holliday, E.G.; Maguire, J.M.; Evans, T.-J.; Koblar, S.A.; Jannes, J.; Sturm, J.W.; Hankey, G.J.; Baker, R.; Golledge, J.; Parsons, M.W.; et al. Common variants at 6p21.1 are associated with large artery atherosclerotic stroke. Nat. Genet. 2012, 44, 1147–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yiallouros, P.K.; Kouis, P.; Pirpa, P.; Michailidou, K.; Loizidou, M.A.; Potamiti, L.; Kalyva, M.; Koutras, G.; Kyriacou, K.; Hadjisavvas, A. Wide phenotypic variability in RSPH9-associated primary ciliary dyskinesia: Review of a case-series from Cyprus. J. Thorac. Dis. 2019, 11, 2067–2075. [Google Scholar] [CrossRef]
- Sherva, R.; Tripodis, Y.; Bennett, D.A.; Chibnik, L.B.; Crane, P.K.; de Jager, P.L.; Farrer, L.A.; Saykin, A.J.; Shulman, J.M.; Naj, A.; et al. Genome-wide association study of the rate of cognitive decline in Alzheimer’s disease. Alzheimer’s Dement. 2014, 10, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Steffens, M.; Leu, C.; Ruppert, A.-K.; Zara, F.; Striano, P.; Robbiano, A.; Capovilla, G.; Tinuper, P.; Gambardella, A.; Bianchi, A.; et al. Genome-wide association analysis of genetic generalized epilepsies implicates susceptibility loci at 1q43, 2p16.1, 2q22.3 and 17q21.32. Hum. Mol. Genet. 2012, 21, 5359–5372. [Google Scholar] [CrossRef]
- Onoufriadis, A.; Shoemark, A.; Munye, M.M.; James, C.T.; Schmidts, M.; Patel, M.; Rosser, E.M.; Bacchelli, C.; Beales, P.L.; Scambler, P.J.; et al. Combined exome and whole-genome sequencing identifies mutations in ARMC4 as a cause of primary ciliary dyskinesia with defects in the outer dynein arm. J. Med. Genet. 2014, 51, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Loth, D.W.; Soler Artigas, M.; Gharib, S.A.; Wain, L.V.; Franceschini, N.; Koch, B.; Pottinger, T.D.; Smith, A.V.; Duan, Q.; Oldmeadow, C.; et al. Genome-wide association analysis identifies six new loci associated with forced vital capacity. Nat. Genet. 2014, 46, 669–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, H.-C.; Li, S.-C.; Guo, M.M.-H.; Huang, Y.-H.; Yu, H.-R.; Huang, F.-C.; Jiao, F.; Kuo, H.-C.; Andrade, J.; Chan, W.-C. Genome-Wide Association Study Identifies Novel Susceptibility Genes Associated with Coronary Artery Aneurysm Formation in Kawasaki Disease. PLoS ONE 2016, 11, e0154943. [Google Scholar] [CrossRef] [Green Version]
- Wiemels, J.L.; Walsh, K.M.; de Smith, A.J.; Metayer, C.; Gonseth, S.; Hansen, H.M.; Francis, S.S.; Ojha, J.; Smirnov, I.; Barcellos, L.; et al. GWAS in childhood acute lymphoblastic leukemia reveals novel genetic associations at chromosomes 17q12 and 8q24.21. Nat. Commun. 2018, 9, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Li, L.; Hu, Z.; Li, S.; Wang, S.; Liu, J.; Wu, C.; He, L.; Zhou, J.; Li, Z.; et al. A genome-wide association study identifies two new cervical cancer susceptibility loci at 4q12 and 17q12. Nat. Genet. 2013, 45, 918–922. [Google Scholar] [CrossRef]
- van der Valk, R.J.; Duijts, L.; Timpson, N.J.; Salam, M.T.; Standl, M.; Curtin, J.A.; Genuneit, J.; Kerhof, M.; Kreiner-Møller, E.; Cáceres, A.; et al. Fraction of exhaled nitric oxide values in childhood are associated with 17q11.2-q12 and 17q12-q21 variants. J. Allergy Clin. Immunol. 2014, 134, 46–55. [Google Scholar] [CrossRef] [Green Version]
- Endeman, H.; Meijvis, S.C.A.; Rijkers, G.T.; van Velzen-Blad, H.; van Moorsel, C.H.M.; Grutters, J.C.; Biesma, D.H. Systemic cytokine response in patients with community-acquired pneumonia. Eur. Respir. J. 2011, 37, 1431–1438. [Google Scholar] [CrossRef] [Green Version]
- Dias, S.; Boyd, R.; Balkwill, F. IL-12 regulates VEGF and MMPs in a murine breast cancer model. Int. J. Cancer 1998, 78, 361–365. [Google Scholar] [CrossRef]
- Ali, M.; Mali, V.; Haddox, S.; AbdelGhany, S.M.; El-Deek, S.E.M.; Abulfadl, A.; Matrougui, K.; Belmadani, S. Essential Role of IL-12 in Angiogenesis in Type 2 Diabetes. Am. J. Pathol. 2017, 187, 2590–2601. [Google Scholar] [CrossRef] [Green Version]
- O’Garra, A.; Murphy, K.M. From IL-10 to IL-12: How pathogens and their products stimulate APCs to induce T(H)1 development. Nat. Immunol. 2009, 10, 929–932. [Google Scholar] [CrossRef] [PubMed]
- Debette, S.; Visvikis-Siest, S.; Chen, M.-H.; Ndiaye, N.-C.; Song, C.; Destefano, A.; Safa, R.; Azimi Nezhad, M.; Sawyer, D.; Marteau, J.-B.; et al. Identification of cis- and trans-acting genetic variants explaining up to half the variation in circulating vascular endothelial growth factor levels. Circ. Res. 2011, 109, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Rouhani, F.N.; Brantly, M.L.; Markello, T.C.; Helip-Wooley, A.; O’Brien, K.; Hess, R.; Huizing, M.; Gahl, W.A.; Gochuico, B.R. Alveolar macrophage dysregulation in Hermansky-Pudlak syndrome type 1. Am. J. Respir. Crit. Care Med. 2009, 180, 1114–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borish, L.; Mascali, J.J.; Dishuck, J.; Beam, W.R.; Martin, R.J.; Rosenwasser, L.J. Detection of alveolar macrophage-derived IL-1 beta in asthma. Inhibition with corticosteroids. J. Immunol. 1992, 149, 3078–3082. [Google Scholar]
- Nishimoto, A.T.; Rosch, J.W.; Tuomanen, E.I. Pneumolysin: Pathogenesis and Therapeutic Target. Front. Microbiol. 2020, 11, 1543. [Google Scholar] [CrossRef]
- Voevodin, A.A.; Khadartsev, A.A.; Bondar, S.S.; Terekhov, I.V.; Nikiforov, V.S. The State of Intracellular Molecular Regulators during the Reconvalescence of Community-Acquired Pneumonia under the Influence of Microwaves at 1 GHz. Integr. Med. Int. 2019, 4, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Hjeij, R.; Lindstrand, A.; Francis, R.; Zariwala, M.A.; Liu, X.; Li, Y.; Damerla, R.; Dougherty, G.W.; Abouhamed, M.; Olbrich, H.; et al. ARMC4 mutations cause primary ciliary dyskinesia with randomization of left/right body asymmetry. Am. J. Hum. Genet. 2013, 93, 357–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castleman, V.H.; Romio, L.; Chodhari, R.; Hirst, R.A.; de Castro, S.C.P.; Parker, K.A.; Ybot-Gonzalez, P.; Emes, R.D.; Wilson, S.W.; Wallis, C.; et al. Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am. J. Hum. Genet. 2009, 84, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Zariwala, M.A.; Gee, H.Y.; Kurkowiak, M.; Al-Mutairi, D.A.; Leigh, M.W.; Hurd, T.W.; Hjeij, R.; Dell, S.D.; Chaki, M.; Dougherty, G.W.; et al. ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6. Am. J. Hum. Genet. 2013, 93, 336–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Trait | Statistics |
|---|---|
| Age [years] | 62 (18–94) |
| Males/Females | 239 (60%)/161 (40%) |
| Body mass index [kg/m2] | 26.3 (15.0–54.3) |
| Current smoking | 117 (29%) |
| Years of smoking (current and former smoker) | 14 (0–65) |
| Years of smoking (only current smoker) | 25 (4–60) |
| Chronic kidney disease | 39 (10%) |
| Chronic liver disease | 9 (2%) |
| Diabetes | 83 (21%) |
| Antibiotic therapy prior to hospitalisation | 100 (25%) |
| Locus | Lead SNP | Cytoband | Physical Position | Physical Nearby Genes (Distance to Lead SNP (kb)) | Best Associated Trait | Effect Allele | Other Allele | Allele Frequency | Beta | SE | p-Value | #SNPs in Credible Set (First: 95%, Second: 99%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| #1 | rs116606423 | 2p16.3 | 50123457 | RPL7P13 (17), NRXN1 (22) | IL-1β | G | T | 0.02 | 1.39 | 0.25 | 3.46 × 10−8 | 3484 3819 |
| #2 | rs139453626 | 3p21.31 | 47700006 | SMARCC1 (0), CSPG5 (78), RN7SL870P (98), DHX30 (140) | IL-10 | G | A | 0.02 | 2.42 | 0.42 | 2.39 × 10−8 | - |
| #3 | rs7763440 | 6p21.1 | 43926708 | C6orf223 (42), MRPL14 (150), TMEM63B (170), VEGFA (170) | VEGF | A | G | 0.45 | −0.65 | 0.07 | 1.58 × 10−20 | 10 12 |
| #4 | rs145122044 | 7q11.23 | 76485397 | UPK3B (0), FDPSP7 (110), DTX2P1 (120), DTX2P1-UPK3BP1-PMS2P11 (120) | MIP-1α (CCL3) | C | T | 0.01 | 1.54 | 0.27 | 1.55 × 10−8 | 3230 3631 |
| #5 | rs62505830 | 8p12 | 36121891 | RN7SKP201 (2.1), MTND6P19 (15), RNU6-533P (45) | IL-1β | T | C | 0.01 | 1.72 | 0.30 | 2.55 × 10−8 | 814 944 |
| #6 | rs36002018 | 9q34.12 | 133656053 | ABL1 (0), EXOSC2 (76), PRDM12 (98), QRFP (110) | IL-10 | T | A | 0.02 | 1.75 | 0.31 | 4.52 × 10−8 | 2534 3160 |
| #7 | rs75237116 | 10p13 | 12655293 | CAMK1D (0), MIR4480 (34), MIR548Q (110), RNU6ATAC39P (160) | MIP-1α (CCL3) | T | C | 0.01 | 1.64 | 0.27 | 2.28 × 10−9 | 3945 4541 |
| #8 | rs6481492 | 10p12.1 | 28207393 | ARMC4 (0), RPL36AP55 (13), MPP7 (130), RN7SKP132 (130) | IL-1β | C | T | 0.06 | 0.79 | 0.13 | 2.76 × 10−9 | 29 50 |
| #9 | rs11223001 | 11q25 | 132161625 | NTM (0), NTM-IT (6.6), OPCML (120), RNU6-1182P (230) | MIP-1α (CCL3) | G | A | 0.01 | 1.56 | 0.27 | 1.73 × 10−8 | - |
| #10 | rs118008913 | 15q14 | 39036672 | C15orf53 (44), RASGRP1 (180) | MIP-1α (CCL3) | G | A | 0.02 | 1.10 | 0.20 | 4.81 × 10−8 | 3397 3761 |
| #11 | rs117439842 | 17q21.32 | 45933872 | SP6 (0.63), SCRN2 (15), LRRC46 (19), MRPL10 (25), OSBPL7 (35), SP2 (40) | IL-1β | T | G | 0.01 | 1.96 | 0.32 | 1.51 × 10−9 | 10 23 |
| #12 | rs8082167 | 17q22 | 52494156 | ISCA1P3 (61) | MIP-1α (CCL3) | T | C | 0.02 | 1.31 | 0.23 | 1.22 × 10−8 | 2838 3247 |
| #13 | rs76920584 | 18q21.2 | 51386971 | - | IL-1β | C | T | 0.01 | 1.49 | 0.26 | 2.11 × 10−8 | 194 1687 |
| #14 | rs3788792 | Xq13.1 | 71585878 | HDAC8 (0), RNU2-68P (11), CITED1 (59), PIN4 (63) | MIP-1α (CCL3) | T | C | 0.02 | 1.17 | 0.19 | 2.74 × 10−9 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kühnapfel, A.; Horn, K.; Klotz, U.; Kiehntopf, M.; Rosolowski, M.; Loeffler, M.; Ahnert, P.; Suttorp, N.; Witzenrath, M.; Scholz, M. Genetic Regulation of Cytokine Response in Patients with Acute Community-Acquired Pneumonia. Genes 2022, 13, 111. https://doi.org/10.3390/genes13010111
Kühnapfel A, Horn K, Klotz U, Kiehntopf M, Rosolowski M, Loeffler M, Ahnert P, Suttorp N, Witzenrath M, Scholz M. Genetic Regulation of Cytokine Response in Patients with Acute Community-Acquired Pneumonia. Genes. 2022; 13(1):111. https://doi.org/10.3390/genes13010111
Chicago/Turabian StyleKühnapfel, Andreas, Katrin Horn, Ulrike Klotz, Michael Kiehntopf, Maciej Rosolowski, Markus Loeffler, Peter Ahnert, Norbert Suttorp, Martin Witzenrath, and Markus Scholz. 2022. "Genetic Regulation of Cytokine Response in Patients with Acute Community-Acquired Pneumonia" Genes 13, no. 1: 111. https://doi.org/10.3390/genes13010111
APA StyleKühnapfel, A., Horn, K., Klotz, U., Kiehntopf, M., Rosolowski, M., Loeffler, M., Ahnert, P., Suttorp, N., Witzenrath, M., & Scholz, M. (2022). Genetic Regulation of Cytokine Response in Patients with Acute Community-Acquired Pneumonia. Genes, 13(1), 111. https://doi.org/10.3390/genes13010111