
Investigation and Management of Apparently Sporadic Central Nervous System Haemangioblastoma for Evidence of Von Hippel–Lindau Disease

 , and
, and
Abstract
:1. Introduction
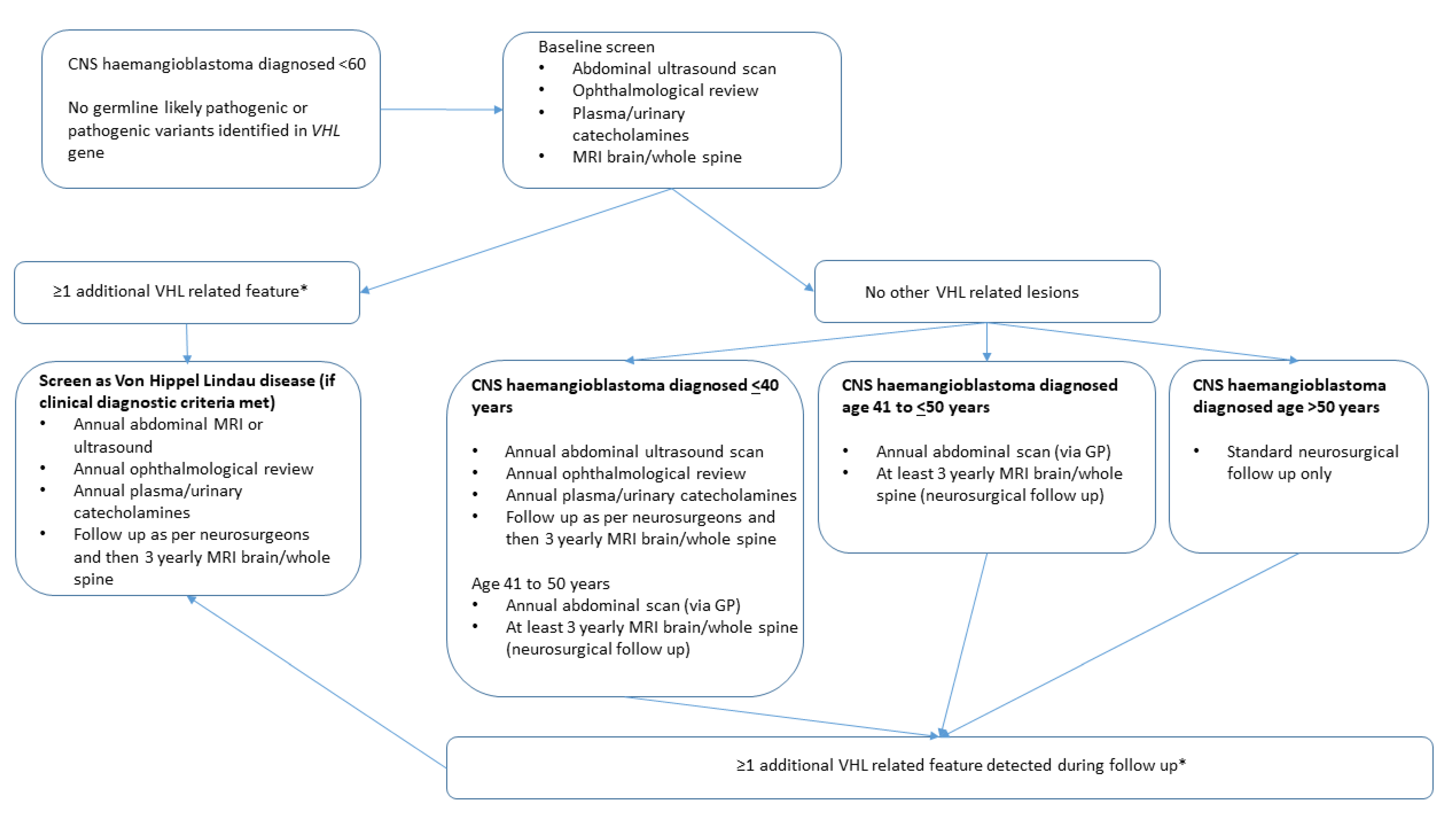
2. Methods
2.1. Audit of Current Practice of Follow-Up for Sporadic CNS Haemangioblastomas
2.2. Audit of Surveillance Results in Individuals with Sporadic CNS Haemangioblastoma and Negative VHL Gene Testing
3. Results
3.1. Audit of Current Practice of Follow-Up for Sporadic CNS Haemangioblastomas in UK Clinical Genetic Centres
3.2. Audit of Surveillance Results in 91 Individuals with Sporadic CNS Haemangioblastoma and Negative VHL Gene Testing
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hussein, M.R. Central nervous system capillary haemangioblastoma: The pathologist’s viewpoint. Int. J. Exp. Pathol. 2007, 88, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.S.; Doan, N.B.; Gelsomino, M.; Shabani, S.; Awad, A.J.; Kaushal, M.; Mortazavi, M.M. Intracranial hemangioblastoma—A SEER-based analysis 2004–2013. Oncotarget 2018, 9, 28009–28015. [Google Scholar] [CrossRef] [Green Version]
- Vortmeyer, A.O.; Gnarra, J.R.; Emmert-Buck, M.R.; Katz, D.; Linehan, W.M.; Oldfield, E.H.; Zhuang, Z. von Hippel-Lindau gene deletion detected in the stromal cell component of a cerebellar hemangioblastoma associated with von Hippel-Lindau disease. Hum. Pathol. 1997, 28, 540–543. [Google Scholar] [CrossRef]
- Maher, E.R.; Yates, J.R.; Ferguson-Smith, M.A. Statistical analysis of the two stage mutation model in von Hippel-Lindau disease, and in sporadic cerebellar haemangioblastoma and renal cell carcinoma. J. Med. Genet. 1990, 27, 311–314. [Google Scholar] [CrossRef] [Green Version]
- Maher, E.R.; Neumann, H.P.; Richard, S. Von Hippel-Lindau disease: A clinical and scientific review. Eur. J. Hum. Genet. 2011, 19, 617–623. [Google Scholar] [CrossRef] [Green Version]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Molecular analysis of de novo germline mutations in the von Hippel-Lindau disease gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Richards, F.M.; Payne, S.J.; Zbar, B.; Affara, N.A.; Ferguson-Smith, M.A.; Maher, E.R. Molecular analysis of de novo germline mutations in the von Hippel-Lindau disease gene. Hum. Mol. Genet. 1995, 4, 2139–2143. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.R.; Yates, J.R.; Harries, R.; Benjamin, C.; Harris, R.; Moore, A.T.; Ferguson-Smith, M.A. Clinical features and natural history of von Hippel-Lindau disease. Q. J. Med. 1990, 77, 1151–1163. [Google Scholar] [CrossRef]
- Hes, F.J.; McKee, S.; Taphoorn, M.J.B.; Rehal, P.; van Der Luijt, R.B.; McMahon, R.; van der Smagtf, J.J.; Dowe, D.; Zewalda, R.A.; Whittaker, J.; et al. Cryptic von Hippel-Lindau disease: Germline mutations in haemangioblastoma-only patients. J. Med. Genet. 2000, 37, 939–943. [Google Scholar] [CrossRef]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumour suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef]
- Melmon, K.L.; Rosen, S.W. Lindau’s disease: Review of the literature and study of a large kindred. Am. J. Med. 1964, 36, 595–617. [Google Scholar] [CrossRef]
- Woodward, E.R.; Wall, K.; Forsyth, J.; Macdonald, F.; Maher, E.R. VHL mutation analysis in patients with isolated central nervous system haemangioblastoma. Brain 2007, 130, 836–842. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Bennett, R.L.; Buchanan, A.; Pearlman, R.; Wiesner, G.L. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet. Med. 2015, 17, 70–87. [Google Scholar] [CrossRef] [Green Version]
- Sgambati, M.T.; Stolle, C.; Choyke, P.L.; Walther, M.M.; Zbar, B.; Linehan, W.M.; Glenn, G.M. Mosaicism in von Hippel-Lindau disease: Lessons from kindreds with germline mutations identified in offspring with mosaic parents. Am. J. Hum. Genet. 2000, 66, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Coppin, L.; Grutzmacher, C.; Crepin, M.; Destailleur, E.; Giraud, S.; Cardot-Bauters, C.; Porchet, N.; Pigny, P. VHL mosaicism can be detected by clinical next-generation sequencing and is s not restricted to patients with a mild phenotype. Eur. J. Hum. Genet. 2014, 22, 1149–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenglet, M.; Robriquet, F.; Schwarz, K.; Camps, C.; Couturier, A.; Hoogewijs, D.; Buffet, A.; Knight, S.J.L.; Gad, S.; Couvé, S.; et al. Identification of a new VHL exon and complex splicing alterations in familial erythrocytosis or von Hippel-Lindau disease. Blood 2018, 132, 469–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordeuk, V.R.; Sergueeva, A.I. Congenital disorder of oxygen sensing: Association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood 2004, 103, 3924–3932. [Google Scholar] [CrossRef] [PubMed]
- Couvé, S.; Ladroue, C.; Laine, E.; Mahtouk, K.; Guégan, J.; Gad, S.; Jeune Le, H.; Gentil Le, M.; Nuel, G.; Kim, W.Y.; et al. Genetic evidence of a precisely tuned dysregulation in the hypoxia signaling pathway during oncogenesis. Cancer Res. 2014, 74, 6554–6564. [Google Scholar] [CrossRef] [Green Version]
- Prowse, A.H.; Webster, A.R.; Richards, F.M.; Richard, S.; Olschwang, S.; Resche, F.; Affara, N.A.; Maher, E.R. Somatic inactivation of the VHL gene in Von Hippel-Lindau disease tumors. Am. J. Hum. Genet. 1997, 60, 765–771. [Google Scholar] [PubMed]
- Lee, J.Y.; Dong, S.M.; Park, W.S.; Yoo, N.J.; Kim, C.S.; Jang, J.J.; Chi, J.-G.; Zbar, B.; Lubensky, I.A.; Marston, W.; et al. Linehan. Loss of heterozygosity and somatic mutations of the VHL tumor suppressor gene in sporadic cerebellar hemangioblastomas. Cancer Res. 1998, 58, 504–508. [Google Scholar]
- Shankar, G.M.; Taylor-Weiner, A.; Lelic, N.; Jones, R.T.; Kim, J.C.; Francis, J.M.; Abedalthagafi, M.; Borges, L.F.; Coumans, J.-V.; Curry, W.T.; et al. Sporadic hemangioblastomas are characterized by cryptic VHL inactivation. Acta Neuropathol. Commun. 2014, 2, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayanagi, S.; Mukasa, A.; Tanaka, S.; Nomura, M.; Omata, M.; Yanagisawa, S.; Yamamoto, S.; Ichimura, K.; Nakatomi, H.; Ueki, K.; et al. Differences in genetic and epigenetic alterations between von Hippel-Lindau disease-related and sporadic hemangioblastomas of the central nervous system. Neuro-Oncol. 2017, 19, 1228–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Patient | Age at Diagnosis of Haemangioblastoma | Total Follow-Up Time (in Years) | Clinical Feature | Age at Detection |
|---|---|---|---|---|
| HAB1 | 47 | 3 | Solitary renal cyst | 48 |
| HAB2 | 43 | 6 | Solitary renal cyst | 43 |
| HAB3 | 18 | 40 | Recurrence HAB | 58 |
| HAB4 | 30 | 2 | Solitary renal cyst | 32 |
| HAB5 | 30 | 2 | Recurrence HAB | 31 |
| Solitary renal cyst | 31 | |||
| HAB6 | 27 | 12 | Multiple renal cysts | 31 |
| HAB7 | 46 | 14 | RCC (4 cm) | 47 |
| Solitary renal cyst | 58 | |||
| HAB8 | 41 | 7 | Multiple renal cysts | 41 |
| HAB9 | 37 | 8 | Recurrence HAB | 42 |
| HAB10 | 37 | 20 | Recurrence HAB | 43 |
| HAB11 | 36 | 8 | Recurrence HAB | 37 |
| HAB12 | 48 | 8 | Solitary renal cyst | 49 |
| HAB13 | 53 | 4 | Multiple renal cysts | 55 |
| HAB14 | 41 | 4 | Multiple renal cysts | 42 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furness, H.; Salfity, L.; Devereux, J.; Halliday, D.; Hanson, H.; Ruddy, D.M.; UK VHL Study Group; Shah, N.; Sultana, G.; Woodward, E.R.; et al. Investigation and Management of Apparently Sporadic Central Nervous System Haemangioblastoma for Evidence of Von Hippel–Lindau Disease. Genes 2021, 12, 1414. https://doi.org/10.3390/genes12091414
Furness H, Salfity L, Devereux J, Halliday D, Hanson H, Ruddy DM, UK VHL Study Group, Shah N, Sultana G, Woodward ER, et al. Investigation and Management of Apparently Sporadic Central Nervous System Haemangioblastoma for Evidence of Von Hippel–Lindau Disease. Genes. 2021; 12(9):1414. https://doi.org/10.3390/genes12091414
Chicago/Turabian StyleFurness, Hugh, Louay Salfity, Johanna Devereux, Dorothy Halliday, Helen Hanson, Deborah M. Ruddy, UK VHL Study Group, Neha Shah, George Sultana, Emma R. Woodward, and et al. 2021. "Investigation and Management of Apparently Sporadic Central Nervous System Haemangioblastoma for Evidence of Von Hippel–Lindau Disease" Genes 12, no. 9: 1414. https://doi.org/10.3390/genes12091414
APA StyleFurness, H., Salfity, L., Devereux, J., Halliday, D., Hanson, H., Ruddy, D. M., UK VHL Study Group, Shah, N., Sultana, G., Woodward, E. R., Sandford, R. N., Snape, K. M., & Maher, E. R. (2021). Investigation and Management of Apparently Sporadic Central Nervous System Haemangioblastoma for Evidence of Von Hippel–Lindau Disease. Genes, 12(9), 1414. https://doi.org/10.3390/genes12091414