
ATM’s Role in the Repair of DNA Double-Strand Breaks


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
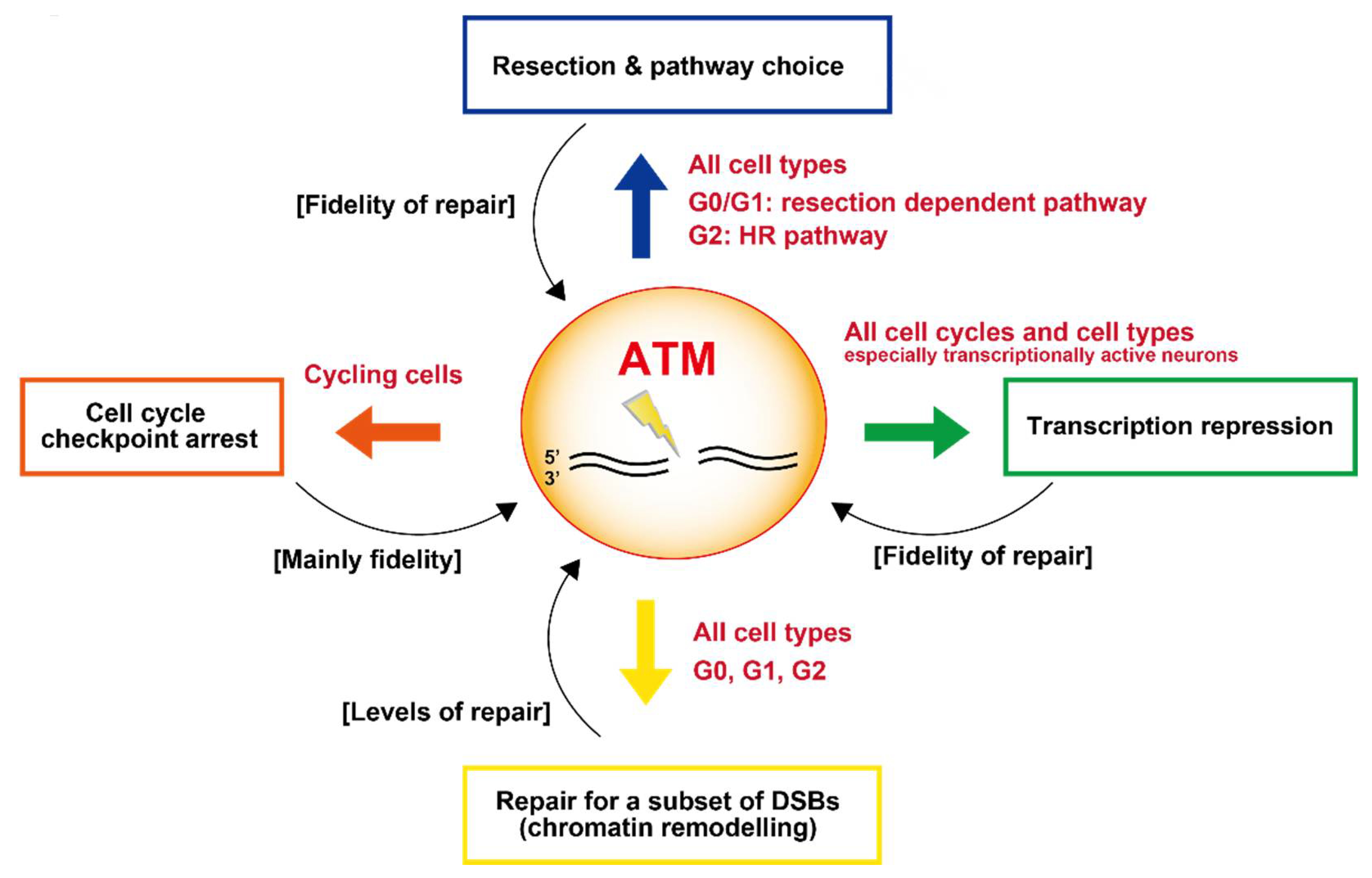
:1. Introduction
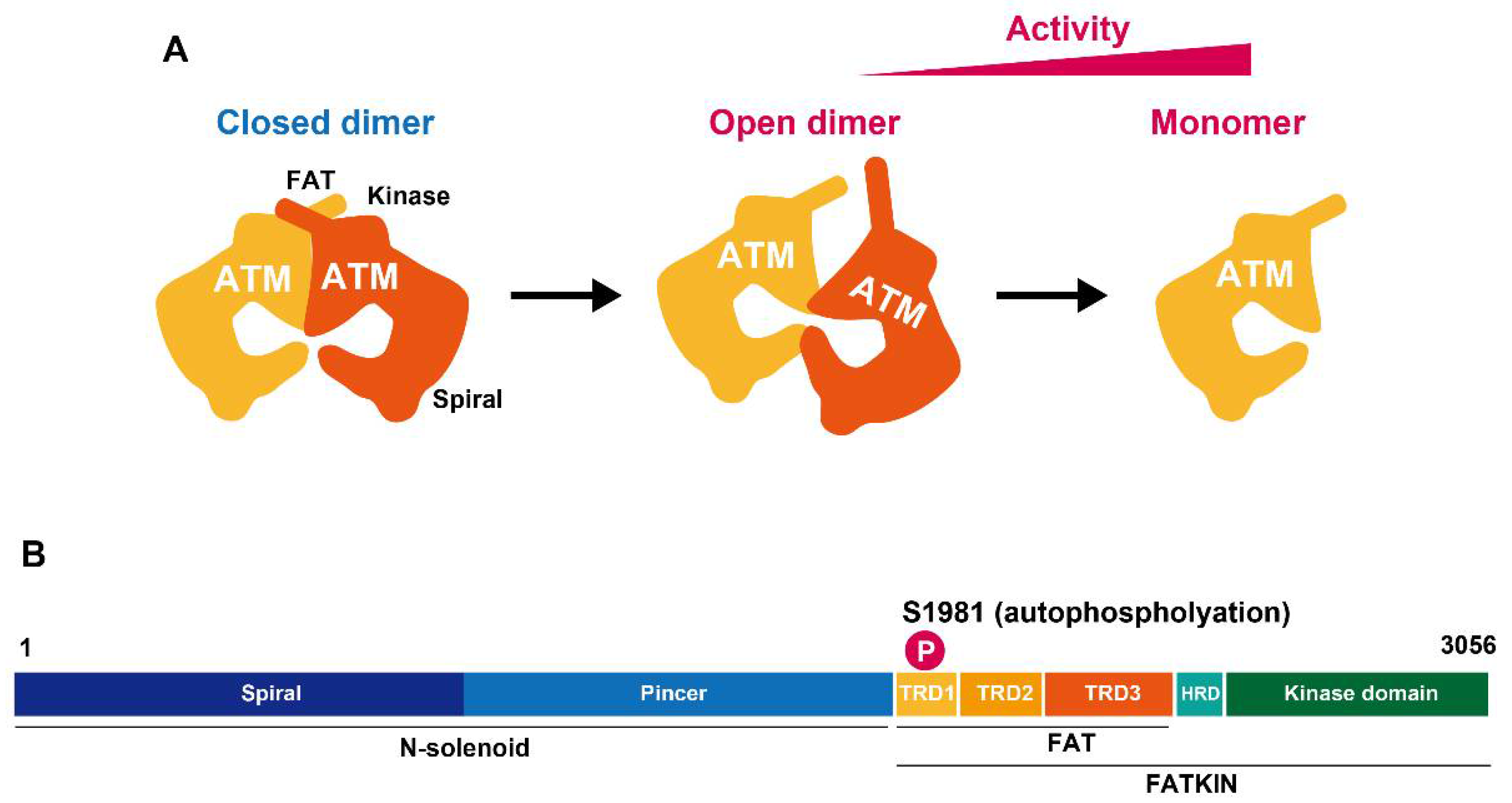
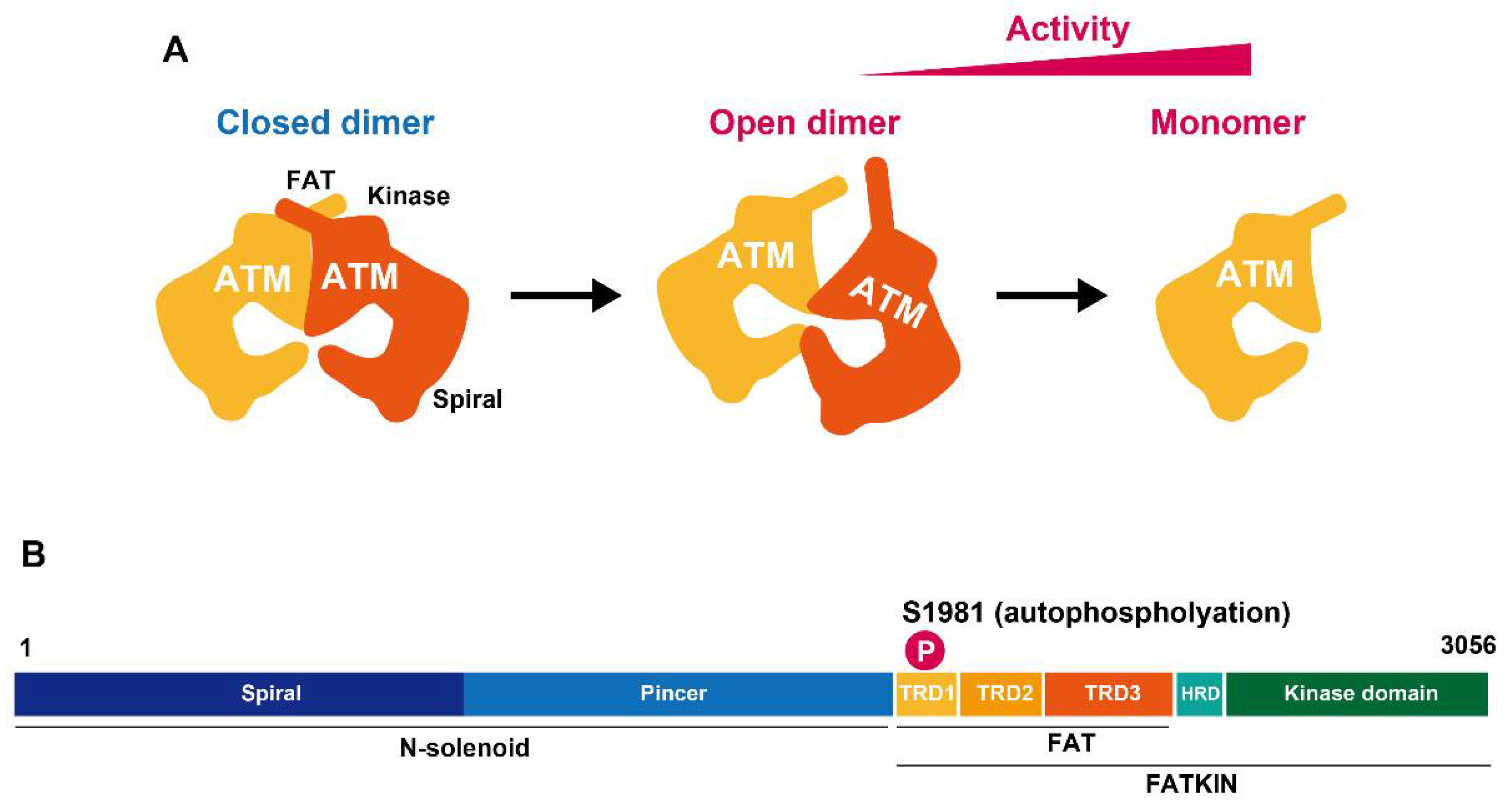
2. ATM Activation
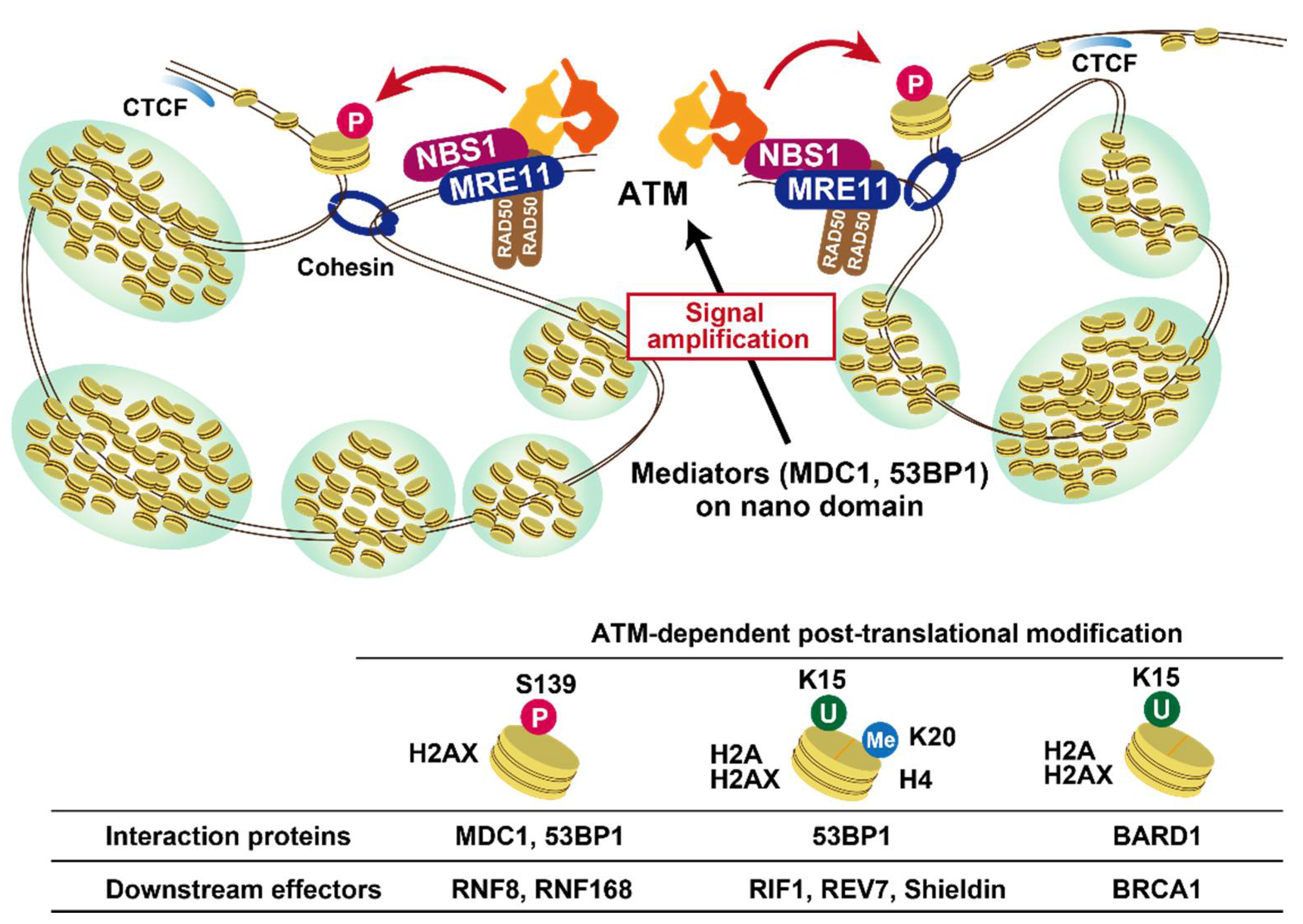
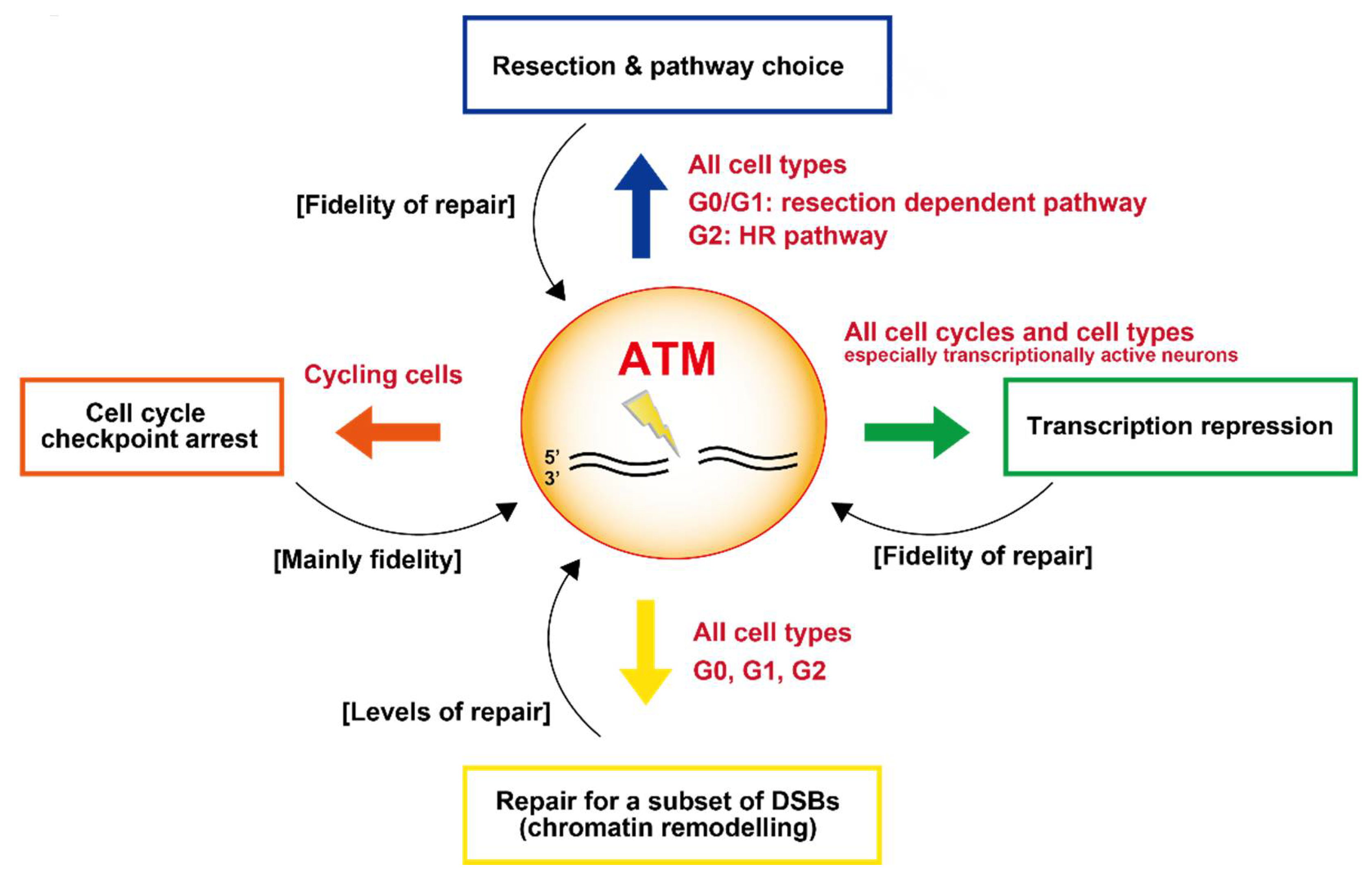
3. Role of ATM in DNA Damage Signalling
4. Cell Cycle Checkpoint Arrest
5. Impact of ATM Signaling on Transcription
6. Role of ATM in the Repair of a Subset of DSBs: Role of Chromatin Remodeling
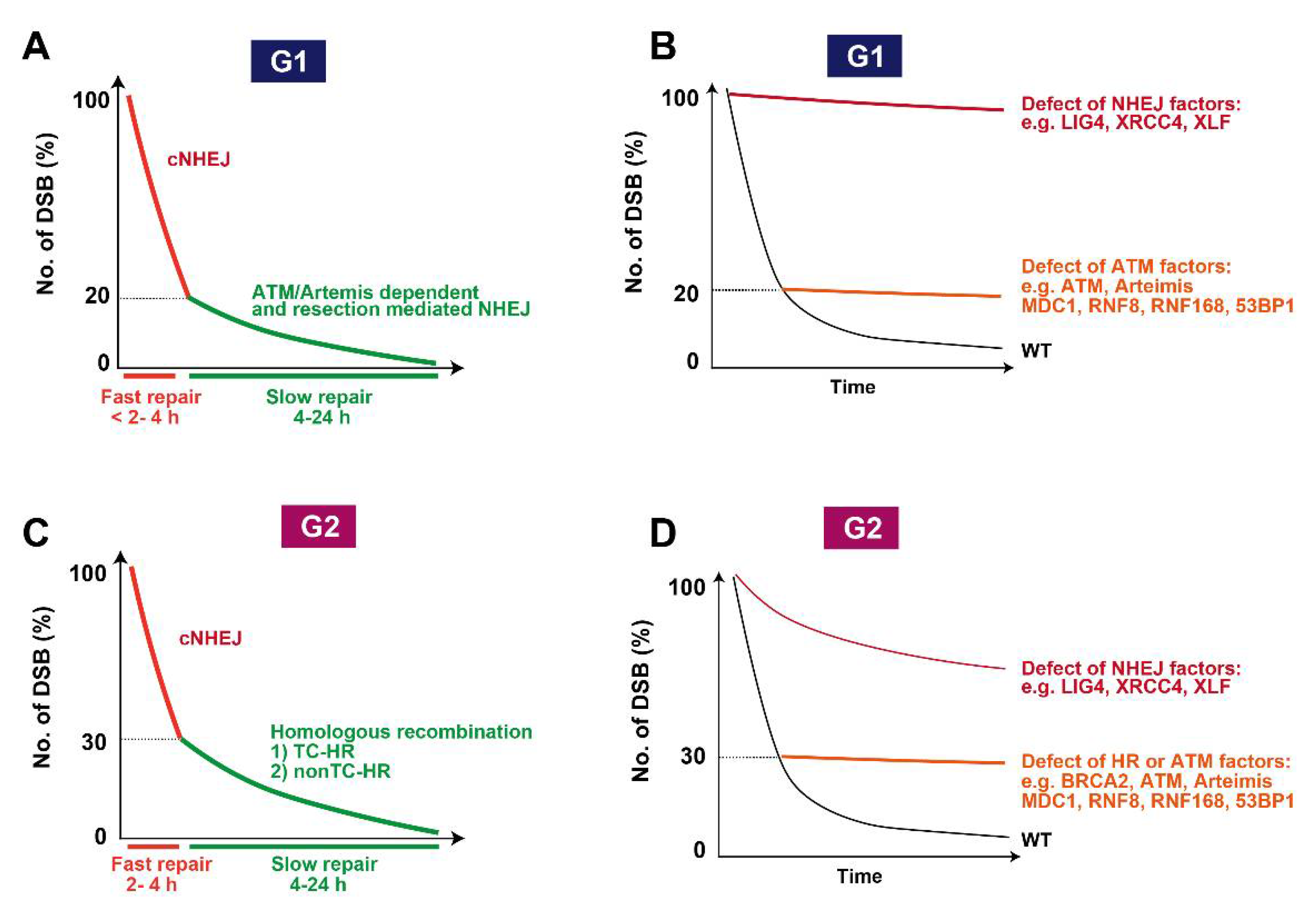
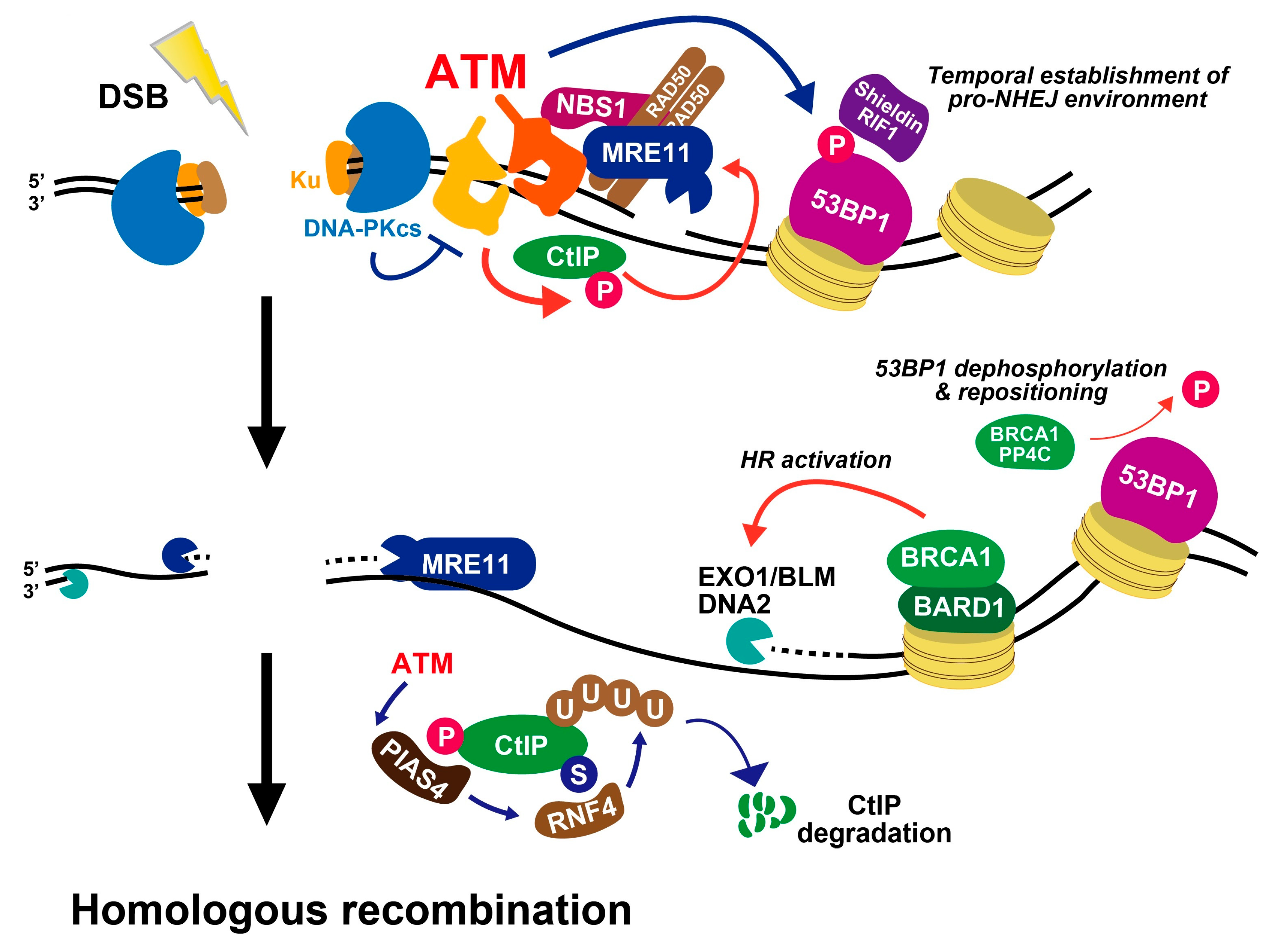
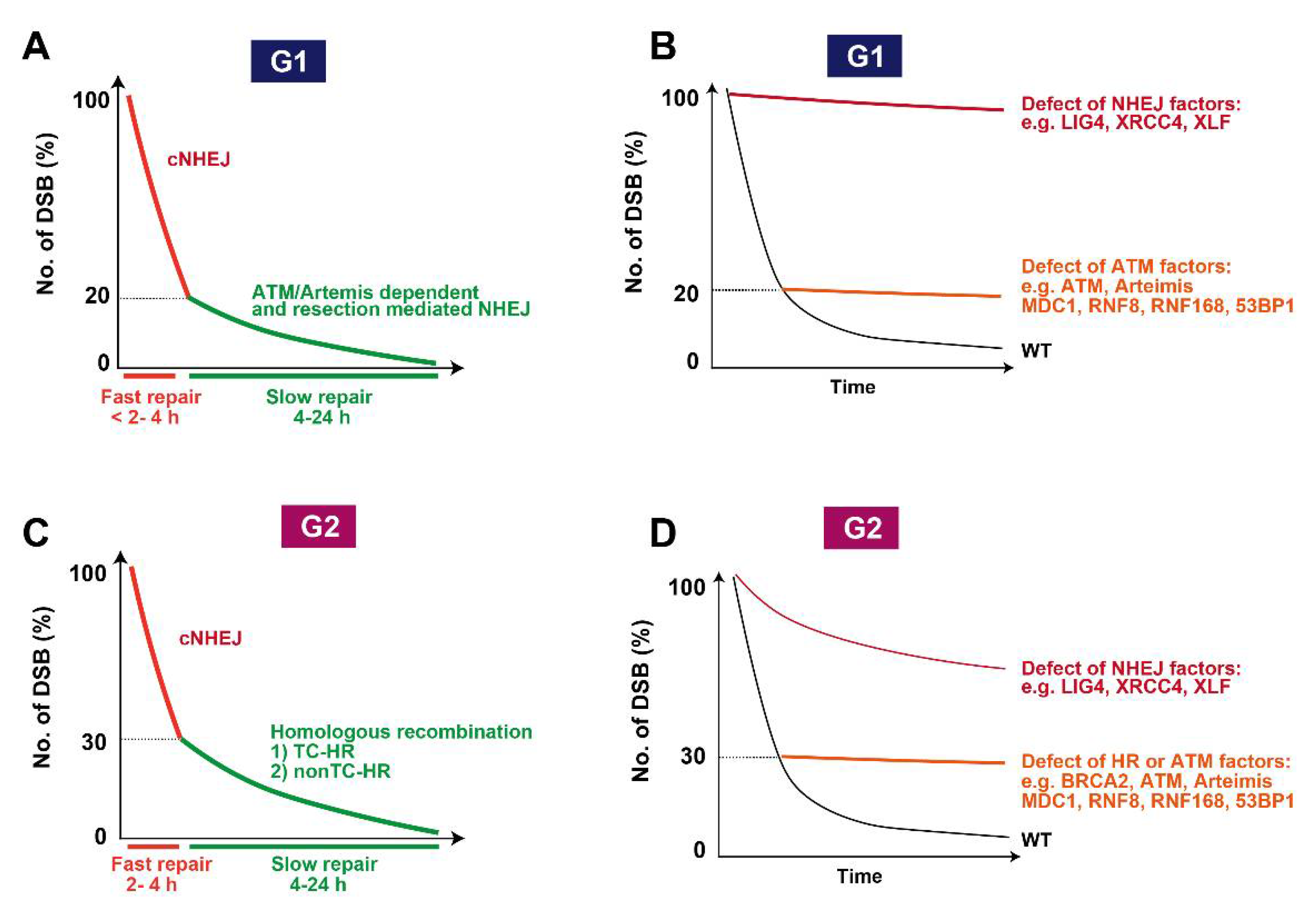
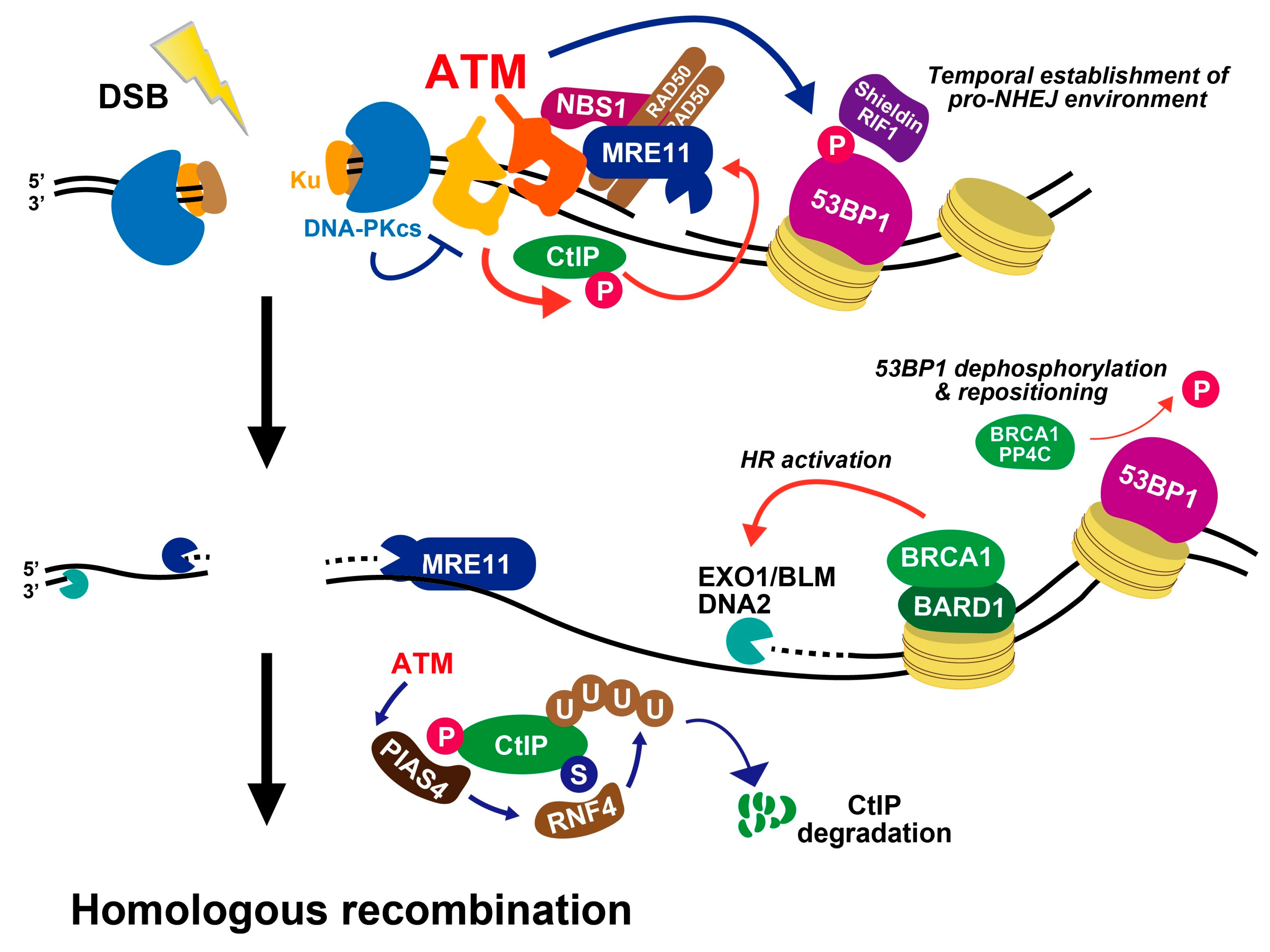
7. Roles of ATM in DSB End Resection Influencing Pathway Choice
8. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, A.M.; Harnden, D.G.; Arlett, C.F.; Harcourt, S.A.; Lehmann, A.R.; Stevens, S.; Bridges, B.A. Ataxia telangiectasia: A human mutation with abnormal radiation sensitivity. Nature 1975, 258, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Lehman, A.R.; Stevens, S. The production and repair of double strand breaks in cells from normal humans and from patients with ataxia telangiectasia. Biochim. Biophys. Acta 1977, 474, 49–60. [Google Scholar] [CrossRef]
- Painter, R.B.; Young, B.R. Radiosensitivity in ataxia-telangiectasia: A new explanation. Proc. Natl. Acad. Sci. USA 1980, 77, 7315–7317. [Google Scholar] [CrossRef] [Green Version]
- Kastan, M.B.; Zhan, Q.; el-Deiry, W.S.; Carrier, F.; Jacks, T.; Walsh, W.V.; Plunkett, B.S.; Vogelstein, B.; Fornace, A.J., Jr. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 1992, 71, 587–597. [Google Scholar] [CrossRef]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef]
- Riballo, E.; Kuhne, M.; Rief, N.; Doherty, A.; Smith, G.C.; Recio, M.J.; Reis, C.; Dahm, K.; Fricke, A.; Krempler, A.; et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol. Cell 2004, 16, 715–724. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Lau, W.C.; Li, Y.; Liu, Z.; Gao, Y.; Zhang, Q.; Huen, M.S. Structure of the human dimeric ATM kinase. Cell Cycle 2016, 15, 1117–1124. [Google Scholar] [CrossRef]
- Baretic, D.; Pollard, H.K.; Fisher, D.I.; Johnson, C.M.; Santhanam, B.; Truman, C.M.; Kouba, T.; Fersht, A.R.; Phillips, C.; Williams, R.L. Structures of closed and open conformations of dimeric human ATM. Sci. Adv. 2017, 3, e1700933. [Google Scholar] [CrossRef] [Green Version]
- Yates, L.A.; Williams, R.M.; Hailemariam, S.; Ayala, R.; Burgers, P.; Zhang, X. Cryo-EM Structure of nucleotide-bound Tel1(ATM) unravels the molecular basis of inhibition and structural rationale for disease-associated mutations. Structure 2020, 28, 96–104.E3. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Liu, M.; Qi, Y.; Chaban, Y.; Gao, C.; Pan, B.; Tian, Y.; Yu, Z.; Li, J.; Zhang, P.; et al. Structural insights into the activation of ATM kinase. Cell Res. 2019, 29, 683–685. [Google Scholar] [CrossRef]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [Green Version]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef]
- Zhou, Y.; Lee, J.H.; Jiang, W.; Crowe, J.L.; Zha, S.; Paull, T.T. Regulation of the DNA damage response by DNA-PKcs inhibitory phosphorylation of ATM. Mol. Cell 2017, 65, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Arnould, C.; Rocher, V.; Finoux, A.L.; Clouaire, T.; Li, K.; Zhou, F.; Caron, P.; Mangeot, P.E.; Ricci, E.P.; Mourad, R.; et al. Loop extrusion as a mechanism for formation of DNA damage repair foci. Nature 2021, 590, 660–665. [Google Scholar] [CrossRef]
- Collins, P.L.; Purman, C.; Porter, S.I.; Nganga, V.; Saini, A.; Hayer, K.E.; Gurewitz, G.L.; Sleckman, B.P.; Bednarski, J.J.; Bassing, C.H.; et al. DNA double-strand breaks induce H2Ax phosphorylation domains in a contact-dependent manner. Nat. Commun. 2020, 11, 3158. [Google Scholar] [CrossRef]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Mand, M.R.; Kao, C.H.; Zhou, Y.; Ryu, S.W.; Richards, A.L.; Coon, J.J.; Paull, T.T. ATM directs DNA damage responses and proteostasis via genetically separable pathways. Sci. Signal. 2018, 11, eaan5598. [Google Scholar] [CrossRef] [Green Version]
- Hadian, K.; Krappmann, D. Signals from the nucleus: Activation of NF-kappaB by cytosolic ATM in the DNA damage response. Sci. Signal. 2011, 4, pe2. [Google Scholar] [CrossRef]
- Shibata, A.; Jeggo, P.A. DNA double-strand break repair in a cellular context. Clin. Oncol. (R. Coll. Radiol.) 2014, 26, 243–249. [Google Scholar] [CrossRef]
- Shibata, A. Regulation of repair pathway choice at two-ended DNA double-strand breaks. Mutat. Res. 2017, 803–805, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Jeggo, P. A historical reflection on our understanding of radiation-induced DNA double strand break repair in somatic mammalian cells; interfacing the past with the present. Int. J. Radiat. Biol. 2019, 95, 945–956. [Google Scholar] [CrossRef] [Green Version]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [Green Version]
- Melander, F.; Bekker-Jensen, S.; Falck, J.; Bartek, J.; Mailand, N.; Lukas, J. Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. J. Cell Biol. 2008, 181, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Jackson, S.P. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep. 2008, 9, 795–801. [Google Scholar] [CrossRef] [Green Version]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Huen, M.S.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007, 131, 901–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Goodarzi, A.A.; Jeggo, P.A.; Paull, T.T. 53BP1 promotes ATM activity through direct interactions with the MRN complex. EMBO J. 2010, 29, 574–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noon, A.T.; Shibata, A.; Rief, N.; Lobrich, M.; Stewart, G.S.; Jeggo, P.A.; Goodarzi, A.A. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat. Cell Biol. 2010, 12, 177–184. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, J. DNA damage checkpoints: From initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007, 19, 238–245. [Google Scholar] [CrossRef]
- Lukas, C.; Falck, J.; Bartkova, J.; Bartek, J.; Lukas, J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat. Cell Biol. 2003, 5, 255–260. [Google Scholar] [CrossRef]
- Fernandez-Capetillo, O.; Chen, H.T.; Celeste, A.; Ward, I.; Romanienko, P.J.; Morales, J.C.; Naka, K.; Xia, Z.; Camerini-Otero, R.D.; Motoyama, N.; et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 2002, 4, 993–997. [Google Scholar] [CrossRef]
- Deckbar, D.; Jeggo, P.A.; Lobrich, M. Understanding the limitations of radiation-induced cell cycle checkpoints. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 271–283. [Google Scholar] [CrossRef] [Green Version]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [Green Version]
- Kruhlak, M.; Crouch, E.E.; Orlov, M.; Montano, C.; Gorski, S.A.; Nussenzweig, A.; Misteli, T.; Phair, R.D.; Casellas, R. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 2007, 447, 730–734. [Google Scholar] [CrossRef]
- Kakarougkas, A.; Ismail, A.; Chambers, A.L.; Riballo, E.; Herbert, A.D.; Kunzel, J.; Lobrich, M.; Jeggo, P.A.; Downs, J.A. Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol. Cell 2014, 55, 723–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meisenberg, C.; Pinder, S.I.; Hopkins, S.R.; Wooller, S.K.; Benstead-Hume, G.; Pearl, F.M.G.; Jeggo, P.A.; Downs, J.A. Repression of transcription at DNA breaks requires cohesin throughout interphase and prevents genome instability. Mol. Cell 2019, 73, 212–223.E7. [Google Scholar] [CrossRef] [Green Version]
- Kuhne, M.; Riballo, E.; Rief, N.; Rothkamm, K.; Jeggo, P.A.; Lobrich, M. A double-strand break repair defect in ATM-deficient cells contributes to radiosensitivity. Cancer Res. 2004, 64, 500–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Lobrich, M.; Jeggo, P.A. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar] [CrossRef]
- Ziv, Y.; Bielopolski, D.; Galanty, Y.; Lukas, C.; Taya, Y.; Schultz, D.C.; Lukas, J.; Bekker-Jensen, S.; Bartek, J.; Shiloh, Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 2006, 8, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Kurka, T.; Jeggo, P.A. KAP-1 phosphorylation regulates CHD3 nucleosome remodeling during the DNA double-strand break response. Nat. Struct. Mol. Biol. 2011, 18, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Jeggo, P.A. The heterochromatic barrier to DNA double strand break repair: How to get the entry visa. Int. J. Mol. Sci. 2012, 13, 11844–11860. [Google Scholar] [CrossRef] [Green Version]
- Baldock, R.A.; Day, M.; Wilkinson, O.J.; Cloney, R.; Jeggo, P.A.; Oliver, A.W.; Watts, F.Z.; Pearl, L.H. ATM Localization and heterochromatin repair depend on direct interaction of the 53BP1-BRCT2 domain with gammaH2AX. Cell Rep. 2015, 13, 2081–2089. [Google Scholar] [CrossRef] [Green Version]
- Hansen, R.K.; Mund, A.; Poulsen, S.L.; Sandoval, M.; Klement, K.; Tsouroula, K.; Tollenaere, M.A.; Raschle, M.; Soria, R.; Offermanns, S.; et al. SCAI promotes DNA double-strand break repair in distinct chromosomal contexts. Nat. Cell Biol. 2016, 18, 1357–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biehs, R.; Steinlage, M.; Barton, O.; Juhasz, S.; Kunzel, J.; Spies, J.; Shibata, A.; Jeggo, P.A.; Lobrich, M. DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol. Cell 2017, 65, 671–684.E5. [Google Scholar] [CrossRef] [Green Version]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef]
- Shiotani, B.; Zou, L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol. Cell 2009, 33, 547–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Lobrich, M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009, 28, 3413–3427. [Google Scholar] [CrossRef] [Green Version]
- Isono, M.; Niimi, A.; Oike, T.; Hagiwara, Y.; Sato, H.; Sekine, R.; Yoshida, Y.; Isobe, S.Y.; Obuse, C.; Nishi, R.; et al. BRCA1 directs the repair pathway to homologous recombination by promoting 53BP1 dephosphorylation. Cell Rep. 2017, 18, 520–532. [Google Scholar] [CrossRef] [Green Version]
- Setiaputra, D.; Durocher, D. Shieldin—The protector of DNA ends. EMBO Rep. 2019, 20, e47560. [Google Scholar] [CrossRef] [PubMed]
- Mirman, Z.; de Lange, T. 53BP1: A DSB escort. Genes Dev. 2020, 34, 7–23. [Google Scholar] [CrossRef] [Green Version]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Cannavo, E.; Cejka, P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 2014, 514, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Ranjha, L.; Cannavo, E.; Cejka, P. Phosphorylated CtIP functions as a co-factor of the MRE11-RAD50-NBS1 endonuclease in DNA end resection. Mol. Cell 2016, 64, 940–950. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Shi, L.Z.; Wong, C.C.; Han, X.; Hwang, P.Y.; Truong, L.N.; Zhu, Q.; Shao, Z.; Chen, D.J.; Berns, M.W.; et al. The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repair. PLoS Genet. 2013, 9, e1003277. [Google Scholar] [CrossRef] [Green Version]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Lobrich, M.; et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef]
- Yasuhara, T.; Kato, R.; Hagiwara, Y.; Shiotani, B.; Yamauchi, M.; Nakada, S.; Shibata, A.; Miyagawa, K. Human Rad52 promotes XPG-mediated R-loop processing to initiate transcription-associated homologous recombination repair. Cell 2018, 175, 558–570.E11. [Google Scholar] [CrossRef] [Green Version]
- Huertas, P.; Jackson, S.P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef] [Green Version]
- Anand, R.; Jasrotia, A.; Bundschuh, D.; Howard, S.M.; Ranjha, L.; Stucki, M.; Cejka, P. NBS1 promotes the endonuclease activity of the MRE11-RAD50 complex by sensing CtIP phosphorylation. EMBO J. 2019, 38, e101005. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Sossick, A.J.; Boulton, S.J.; Jackson, S.P. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci. 2012, 125, 3529–3534. [Google Scholar] [CrossRef] [Green Version]
- Kakarougkas, A.; Ismail, A.; Katsuki, Y.; Freire, R.; Shibata, A.; Jeggo, P.A. Co-operation of BRCA1 and POH1 relieves the barriers posed by 53BP1 and RAP80 to resection. Nucleic Acids Res. 2013, 41, 10298–10311. [Google Scholar] [CrossRef]
- Kakarougkas, A.; Ismail, A.; Klement, K.; Goodarzi, A.A.; Conrad, S.; Freire, R.; Shibata, A.; Lobrich, M.; Jeggo, P.A. Opposing roles for 53BP1 during homologous recombination. Nucleic Acids Res. 2013, 41, 9719–9731. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Wan, L.; Jiang, G.; Cao, L.; Xia, F.; Tian, T.; Zhu, X.; Wu, M.; Huen, M.S.Y.; Wang, Y.; et al. ATM controls the extent of DNA end resection by eliciting sequential posttranslational modifications of CtIP. Proc. Natl. Acad. Sci. USA 2021, 118, e2022600118. [Google Scholar] [CrossRef]
- Bakr, A.; Oing, C.; Kocher, S.; Borgmann, K.; Dornreiter, I.; Petersen, C.; Dikomey, E.; Mansour, W.Y. Involvement of ATM in homologous recombination after end resection and RAD51 nucleofilament formation. Nucleic Acids Res. 2015, 43, 3154–3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jachimowicz, R.D.; Beleggia, F.; Isensee, J.; Velpula, B.B.; Goergens, J.; Bustos, M.A.; Doll, M.A.; Shenoy, A.; Checa-Rodriguez, C.; Wiederstein, J.L.; et al. UBQLN4 represses homologous recombination and is overexpressed in aggressive tumors. Cell 2019, 176, 505–519.E22. [Google Scholar] [CrossRef] [Green Version]
- Balmus, G.; Pilger, D.; Coates, J.; Demir, M.; Sczaniecka-Clift, M.; Barros, A.C.; Woods, M.; Fu, B.; Yang, F.; Chen, E.; et al. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat. Commun. 2019, 10, 87. [Google Scholar] [CrossRef] [Green Version]
- Chanut, P.; Britton, S.; Coates, J.; Jackson, S.P.; Calsou, P. Coordinated nuclease activities counteract Ku at single-ended DNA double-strand breaks. Nat. Commun. 2016, 7, 12889. [Google Scholar] [CrossRef] [Green Version]
- Britton, S.; Chanut, P.; Delteil, C.; Barboule, N.; Frit, P.; Calsou, P. ATM antagonizes NHEJ proteins assembly and DNA-ends synapsis at single-ended DNA double strand breaks. Nucleic Acids Res. 2020, 48, 9710–9723. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shibata, A.; Jeggo, P.A. ATM’s Role in the Repair of DNA Double-Strand Breaks. Genes 2021, 12, 1370. https://doi.org/10.3390/genes12091370
Shibata A, Jeggo PA. ATM’s Role in the Repair of DNA Double-Strand Breaks. Genes. 2021; 12(9):1370. https://doi.org/10.3390/genes12091370
Chicago/Turabian StyleShibata, Atsushi, and Penny A. Jeggo. 2021. "ATM’s Role in the Repair of DNA Double-Strand Breaks" Genes 12, no. 9: 1370. https://doi.org/10.3390/genes12091370
APA StyleShibata, A., & Jeggo, P. A. (2021). ATM’s Role in the Repair of DNA Double-Strand Breaks. Genes, 12(9), 1370. https://doi.org/10.3390/genes12091370