
Clinical, Biochemical, and Genetic Heterogeneity in Glutaric Aciduria Type II Patients

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Consideration and Data Collection
2.2. Computational Analysis of Variants
2.3. Evaluation of the Effect of Missense Variants on the Protein Stability
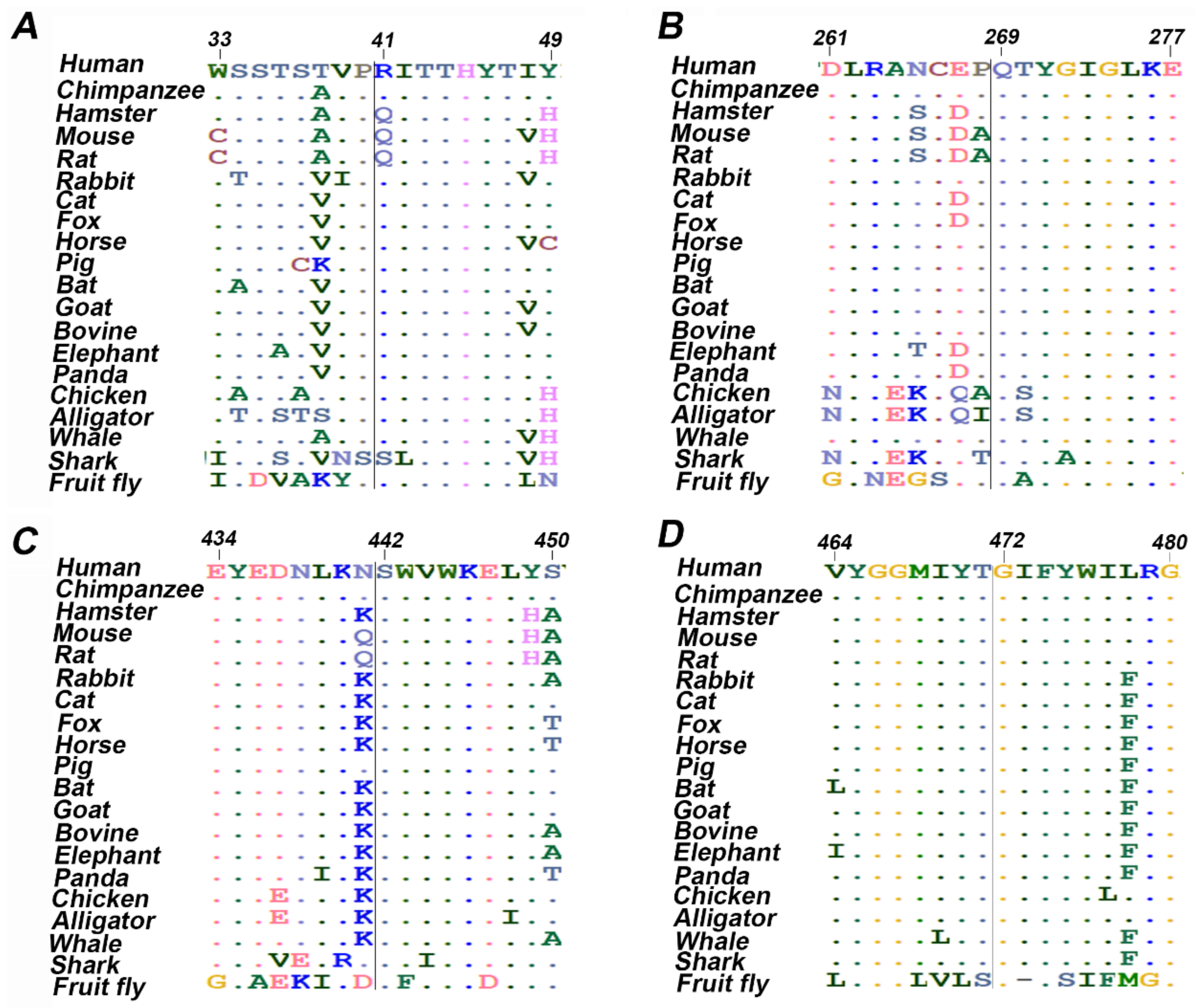
2.4. Evaluation of Protein Evolutionary Conservation
2.5. Evaluation of the Effects of Missense Variants Using Three-Dimensional Protein Modeling
3. Results
3.1. Clinical, Biochemical, and Genetic Characteristics of the Studied Patients
3.2. In Silico Analysis of Identified Variant Pathogenicity
3.3. Evolutionary Conservation Analysis
3.4. Evaluation of The Structural Stability and Effect of Missense Variants on ETF-QO Protein
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vernon, H.J. Inborn Errors of Metabolism: Advances in Diagnosis and Therapy. JAMA Pediatr. 2015, 169, 778–782. [Google Scholar] [CrossRef]
- Ferreira, C.R.; van Karnebeek, C.D.M.; Vockley, J.; Blau, N. A proposed nosology of inborn errors of metabolism. Genet. Med. 2019, 21, 102–106. [Google Scholar] [CrossRef]
- Angle, B.; Burton, B.K. Risk of sudden death and acute life-threatening events in patients with glutaric acidemia type II. Mol. Genet. Metab. 2007, 93, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.I.; Binard, R.J.; Woontner, M.R.; Frerman, F.E. Glutaric acidemia type II: Gene structure and mutations of the electron transfer flavoprotein:ubiquinone oxidoreductase (ETF:QO) gene. Mol. Genet. Metab. 2002, 77, 86–90. [Google Scholar] [CrossRef]
- Shioya, A.; Takuma, H.; Yamaguchi, S.; Ishii, A.; Hiroki, M.; Fukuda, T.; Sugie, H.; Shigematsu, Y.; Tamaoka, A. Amelioration of acylcarnitine profile using bezafibrate and riboflavin in a case of adult-onset glutaric acidemia type 2 with novel mutations of the electron transfer flavoprotein dehydrogenase (ETFDH) gene. J. Neurol. Sci. 2014, 346, 350–352. [Google Scholar] [CrossRef]
- Yamada, K.; Kobayashi, H.; Bo, R.; Takahashi, T.; Purevsuren, J.; Hasegawa, Y.; Taketani, T.; Fukuda, S.; Ohkubo, T.; Yokota, T.; et al. Clinical, biochemical and molecular investigation of adult-onset glutaric acidemia type II: Characteristics in comparison with pediatric cases. Brain Dev. 2016, 38, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Kölker, S.; Christensen, E.; Leonard, J.V.; Greenberg, C.R.; Boneh, A.; Burlina, A.B.; Burlina, A.P.; Dixon, M.; Duran, M.; Cazorla, A.G.; et al. Diagnosis and management of glutaric aciduria type I—Revised recommendations. J. Inherit. Metab. Dis. 2011, 34, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.L.; Frerman, F.E.; Kim, J.-J.P. Three-dimensional structure of human electron transfer flavoprotein to 2.1-Å resolution (X-ray crystallographyglutaric acidemia type II). Proc. Natl. Acad. Sci. USA 1996, 93, 14355–14360. [Google Scholar] [CrossRef]
- Šimkovič, M.; Degala, G.D.; Eaton, S.S.; Frerman, F.E. Expression of human electron transfer flavoprotein-ubiquinone oxidoreductase from a baculovirus vector: Kinetic and spectral characterization of the human protein. Biochem. J. 2002, 364, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Keese, S.M.; Tanaka, K. Biosynthesis of electron transfer flavoprotein in a cell-free system and in cultured human fibroblasts. Defect in the alpha subunit synthesis is a primary lesion in glutaric aciduria type II. J. Clin. Investig. 1986, 78, 997–1002. [Google Scholar] [CrossRef]
- Olsen, R.K.J.; Andresen, B.S.; Christensen, E.; Bross, P.; Skovby, F.; Gregersen, N. Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl-CoA dehydrogenation deficiency. Hum. Mutat. 2003, 22, 12–23. [Google Scholar] [CrossRef]
- Schiff, M.; Froissart, R.; Olsen, R.K.J.; Acquaviva, C.; Vianey-Saban, C. Electron transfer flavoprotein deficiency: Functional and molecular aspects. Mol. Genet. Metab. 2006, 88, 153–158. [Google Scholar] [CrossRef]
- Frerman, F. Defects of electron transfer flavoprotein and electron transfer flavoprotein-ubiquinone oxidoreductase Glutaric aciduria type II. Mol. Genet. Metab. Inherit. Dis. 2001. [Google Scholar] [CrossRef]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human gene mutation database (HGMD): 2003 update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Ou, M.; Zhu, L.; Zhang, Y.; Zhou, J.; Chen, X.; Yang, L.; Li, T.; Su, X.; Zhang, Y.; Zhang, Y.; et al. A novel electron transfer flavoprotein dehydrogenase (ETFDH) gene mutation identified in a newborn with glutaric acidemia type II: A case report of a Chinese family. BMC Med. Genom. 2020, 21, 98. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Wen, B.; Lin, J.; Zhu, W.; Luo, S.; Zhao, C.; Li, D.; Lin, P.; Lu, J.; Yan, C. Clinical features and ETFDH mutation spectrum in a cohort of 90 Chinese patients with late-onset multiple acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2014, 37, 399–404. [Google Scholar] [CrossRef] [PubMed]
- ACT Sheets and Algorithms. Available online: https://www.acmg.net/ACMG/Medical-Genetics-Practice-Resources/ACT_Sheets_and_Algorithms.aspx (accessed on 16 August 2021).
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jhong, J.-H.; Lee, J.; Koo, J.-Y. Meta-analytic support vector machine for integrating multiple omics data. BioData Min. 2017, 10, 1–14. [Google Scholar]
- Zaucha, J.; Heinzinger, M.; Tarnovskaya, S.; Rost, B.; Frishman, D. Family-specific analysis of variant pathogenicity prediction tools. NAR Genom. Bioinform. 2020, 2, lqaa014. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef]
- Gonzalez-Perez, A.; Lopez-Bigas, N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 2011, 88, 440–449. [Google Scholar] [CrossRef]
- Xue, Y.; Zhou, F.; Zhu, M.; Ahmed, K.; Chen, G.; Yao, X. GPS: A comprehensive www server for phosphorylation sites prediction. Nucleic Acids Res. 2005, 33, W184–W187. [Google Scholar] [CrossRef]
- Ross, K.E.; Arighi, C.N.; Ren, J. Construction of Protein Phosphorylation Networks by Data Mining, Text Mining, and Ontology Integration: Analysis of the Spindle Checkpoint. Database 2013, 2013, bat038. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Birney, E.; Biswas, M.; Bucher, P.; Cerutti, L.; Corpet, F.; Croning, M.D.R.; et al. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 2001, 29, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [PubMed]
- Venselaar, H.; Te Beek, T.A.; Kuipers, R.K.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef]
- Buchan, D.W.; Jones, D.T. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 2019, 47, 402–407. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef]
- Capra, J.A.; Singh, M. Predicting functionally important residues from sequence conservation. Bioinformatics 2007, 23, 1875–1882. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System. 2002. Available online: http://www.pymol.org (accessed on 16 August 2021).
- Khan, S.; Vihinen, M. Spectrum of disease-causing mutations in protein secondary structures. BMC Struct. Biol. 2007, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Cang, Z.; Yao, J.; Kim, M.; Deans, E.; Wei, G.; Kang, S.-G.; Hong, H. Structural cavities are critical to balancing stability and activity of a membrane-integral enzyme. Proc. Natl. Acad. Sci. USA 2020, 117, 22146–22156. [Google Scholar] [CrossRef] [PubMed]
- Heydenreich, F.M.; Vuckovic, Z.; Matkovic, M.; Veprintsev, D.B. Stabilization of G protein-coupled receptors by point mutations. Front. Pharmacol. 2015, 6, 82. [Google Scholar] [CrossRef]
- Er, T.K.; Chen, C.C.; Liu, Y.Y.; Chang, H.C.; Chien, Y.H.; Chang, J.G.; Hwang, J.-K.; Jong, Y.-J. Computational analysis of a novel mutation in ETFDH gene highlights its long-range effects on the FAD-binding motif. BMC Struct. Biol. 2011, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Pollard, L.M.; Williams, N.R.; Espinoza, L.; Wood, T.C.; Spector, E.B.; Schroer, R.J.; Holden, K.R. Diagnosis, treatment, and long-term outcomes of late-onset (Type III) multiple Acyl-CoA dehydrogenase deficiency. J. Child. Neurol. 2010, 25, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Wasant, P.; Kuptanon, C.; Vattanavicharn, N.; Liammongkolkul, S.; Ratanarak, P.; Sangruchi, T.; Yamaguchi, S. Glutaric aciduria type 2, late onset type in thai siblings with myopathy. Pediat. Neurol. 2010, 43, 279–282. [Google Scholar] [CrossRef]
- Mareska, M.C.; Adams, K.K.; Muenzer, J.; Frerman, F.; Braun, T.G.; Howard, J.F., Jr. Adult-Onset Presentation of Glutaric Acidemia Type II with Myopathy. J. Clin. Neuromuscul. Dis. 2003, 4, 124–128. [Google Scholar] [CrossRef]
- Russell, A.P.; Schrauwen, P.; Somm, E.; Gastaldi, G.; Hesselink, M.K.C.; Schaart, G.; Kornips, E.; Lo, S.K.; Bufano, D.; Giacobino, J.; et al. Decreased Fatty Acid β-Oxidation in Riboflavin-Responsive, Multiple Acylcoenzyme a Dehydrogenase-Deficient Patients Is Associated with an Increase in Uncoupling Protein-3. J. Clin. Endocrinol. Metab. 2003, 88, 5921–5926. [Google Scholar] [CrossRef][Green Version]
- Wen, B.; Dai, T.; Li, W.; Zhao, Y.; Liu, S.; Zhang, C.; Li, H.; Wu, J.; Li, D.; Yan, C. Riboflavin-responsive lipid-storage myopathy caused by ETFDH gene mutations. J. Neurol. Neurosurg. Psychiatry 2010, 81, 231–236. [Google Scholar] [CrossRef]
- Van Hove, J.L.K.; Grünewald, S.; Jaeken, J.; Demaerel, P.; Declercq, P.E.; Bourdoux, P.; Niezen-Koning, K.; Deanfeld, J.E.; Leonard, J.V. D,L-3-hydroxybutyrate treatment of multiple acyl-CoA dehydrogenase deficiency (MADD). Lancet 2003, 361, 1433–1445. [Google Scholar] [CrossRef]
- Boy, N.; Mengler, K.; Thimm, E.; Schiergens, K.A.; Marquardt, T.; Weinhold, N.; Marquardt, I.; Das, A.M.; Freisinger, P.; Grünert, S.C.; et al. Newborn screening: A disease-changing intervention for glutaric aciduria type 1. Ann. Neurol. 2018, 83, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Couce, M.L.; López-Suárez, O.; Bóveda, M.D.; Castiñeiras, D.E.; Cocho, J.A.; García-Villoria, J.; Castro-Gago, M.; Fraga, J.M.; Ribes, A. Glutaric aciduria type I: Outcome of patients with early-versus late-diagnosis. Eur. J. Paediatr. Neurol. 2013, 17, 383–389. [Google Scholar] [CrossRef]
- Viau, K.; Ernst, S.L.; Vanzo, R.J.; Botto, L.D.; Pasquali, M.; Longo, N. Glutaric acidemia Type 1: Outcomes before and after expanded newborn screening. Mol. Genet. Metab. 2012, 106, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Kolker, S.; Garbade, S.F.; Greenberg, C.R.; Leonard, J.V.; Saudubray, J.M.; Ribes, A.; Kalkanoglu, H.S.; Lund, A.M.; Merinero, B.; Wajner, M.; et al. Natural history, outcome, and treatment efficacy in children and adults with glutaryl-CoA dehydrogenase deficiency. Pediatr. Res. 2006, 59, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Bijarnia, S.; Wiley, V.; Carpenter, K.; Christodoulou, J.; Ellaway, C.J.; Wilcken, B. Glutaric aciduria type I: Outcome following detection by newborn screening. J. Inherit. Metab. Dis. 2008, 31, 503–507. [Google Scholar] [CrossRef]
- Boneh, A.; Beauchamp, M.; Humphrey, M.; Watkins, J.; Peters, H.; Yaplito-Lee, J. Newborn screening for glutaric aciduria type I in Victoria: Treatment and outcome. Mol. Genet. Metab. 2008, 94, 287–291. [Google Scholar] [CrossRef]
- Kölker, S.; Garbade, S.F.; Boy, N.; Maier, E.M.; Meissner, T.; Mühlhausen, C.; Hennermann, J.B.; Lücke, T.; Häberle, J.; Baumkötter, J.; et al. Decline of acute encephalopathic crises in children with glutaryl-CoA dehydrogenase deficiency identified by newborn screening in Germany. Pediatr. Res. 2007, 62, 357–363. [Google Scholar] [CrossRef]
- Burga, A.; Lehner, B. Beyond genotype to phenotype: Why the phenotype of an individual cannot always be predicted from their genome sequence and the environment that they experience. FEBS J. 2012, 279, 3765–3775. [Google Scholar] [CrossRef]
- Clark, A.G. Limits to prediction of phenotypes from knowledge of genotypes. Evol. Biol. 2000, 205–224. [Google Scholar] [CrossRef]
- Law, L.K.; Tang, N.L.; Hui, J.; Fung, S.L.; Ruiter, J.; Wanders, R.J.; Fok, T.-F.; Lam, C.W.K. Novel mutations in ETFDH gene in Chinese patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Clin. Chim. Acta 2009, 404, 95–99. [Google Scholar] [CrossRef]
- Grice, A.; Peck, T. Multiple acyl-CoA dehydrogenase deficiency: A rare cause of acidosis with an increased anion gap. Br. J. Anaesth. 2001, 86, 437–441. [Google Scholar] [CrossRef]
- Lan, M.Y.; Fu, M.H.; Liu, Y.F.; Huang, C.C.; Chang, Y.Y.; Liu, J.S.; Peng, C.-H.; Chen, S.-S. High frequency of ETFDH c.250G>A mutation in Taiwanese patients with late-onset lipid storage myopathy. Clin. Genet. 2010, 78, 565–569. [Google Scholar] [CrossRef]
- Fitzgerald, M.; Crushell, E.; Hickey, C. Cyclic vomiting syndrome masking a fatal metabolic disease. Eur. J. Pediatr. 2013, 172, 707–710. [Google Scholar] [CrossRef]
- Olsen, R.K.J.; Olpin, S.E.; Andresen, B.S.; Miedzybrodzka, Z.H.; Pourfarzam, M.; Merinero, B.; Frerman, F.E.; Beresford, M.W.; Dean, J.C.S.; Cornelius, N.; et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain 2007, 130, 2045–2054. [Google Scholar] [CrossRef]
- Vergani, L.; Barile, M.; Angelini, C.; Burlina, A.B.; Nijtmans, L.; Freda, M.P.; Brizio, C.; Zerbetto, E.; Dabbeni-Sala, F. Riboflavin therapy Biochemical heterogeneity in two adult lipid storage myopathies. Brain 1999, 122, 2401–2411. [Google Scholar] [CrossRef][Green Version]
- Triggs, W.J.; Roe, C.R.; Rhead, W.J.; Hanson, S.K.; Lin, S.-N.; James Willmore, L. Neuropsychiatric manifestations of defect in mitochondrial beta oxidation response to riboflavin the multiple acyl-CoA dehydrogenation disorders (MAD) include glutaric aciduria type II, ethylmalonic-adipic aciduria, and riboflavin-responsive C6-C1O dicarboxylic aciduria.1 The primary defect in MAD seems. J. Neurol. Neurosurg. Psychiatry 1992, 55, 209–230. [Google Scholar]
- Wang, Z.-Q.; Chen, X.-J.; Murong, S.-X.; Wang, N.; Wu, Z.-Y. Molecular analysis of 51 unrelated pedigrees with late-onset multiple acyl-CoA dehydrogenation deficiency (MADD) in southern China confirmed the most common ETFDH mutation and high carrier frequency of c. 250G > A. J. Mol. Med. 2011, 89, 569–576. [Google Scholar] [CrossRef]
- Zhu, M.; Zhu, X.; Qi, X.; Weijiang, D.; Yu, Y.; Wan, H.; Hong, D. Riboflavin-responsive multiple Acyl-CoA dehydrogenation deficiency in 13 cases, and a literature review in mainland Chinese patients. J. Hum. Genet. 2014, 59, 256–261. [Google Scholar] [CrossRef]
- Alves, E.; Henriques, B.J.; Rodrigues, J.V.; Prudêncio, P.; Rocha, H.; Vilarinho, L.; Martinho, R.G.; Gomes, C.M. Mutations at the flavin binding site of ETF:QO yield a MADD-like severe phenotype in Drosophila. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2012, 1822, 1284–1292. [Google Scholar] [CrossRef]
- Ali, A.; Dhahouri, N.A.; Almesmari, F.S.A.; Fathalla, W.M.; Jasmi, F.A. Characterization of ETFDH and PHGDH Mutations in a Patient with Mild Glutaric Aciduria Type II and Serine Deficiency. Genes 2021, 12, 703. [Google Scholar] [CrossRef]
- Deller, M.C.; Kong, L.; Rupp, B. Protein stability: A crystallographer’s perspective. Acta Cryst. 2016, 72, 72–95. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Case I.D | Clinical Phenotype | Gender | Age at Onset | Age at Diagnosis | Parents | Variant | Zygosity | Variant Detection |
|---|---|---|---|---|---|---|---|---|
| 1 | Asymptomatic; no metabolic crises | Male | NA | 2 months | First cousins | ETFDH:c.112G > T p.Arg41Leu; missense | Homozygous | During NBS confirmatory workup |
| 2 | Metabolic crises; encephalopathy; hyperammonemia and metabolic acidosis; recurrent vomiting | Male | 26 years | 26 years | Second cousins | ETFDH:c.807C > T, p.Gln269His; missense | Homozygous | Based on clinical presentation; NBS was not performed |
| 3 | Metabolic crises; cyclic vomiting; abdominal pain, nausea | Female | 20 months | 7 years | First cousins | ETFDH:c.807C > T, p.Gln269His; missense | Homozygous | Based on clinical presentation; NBS was not performed |
| 4 | Metabolic crises; cyclic vomiting; epigastric pain, nausea | Female | 15 years | 5 years | First cousins | ETFDH:c.807C > T, p.Gln269His; missense | Homozygous | As part of Family screening; NBS was not performed |
| 5 | Mild glutaric aciduria type II | Female | 8 years | 5 months | First cousins | ETFDH:c.807C > T, p.Gln269His; missense | Homozygous | As part of Family screening; NBS was not performed |
| 6 | Asymptomatic | Male | 10 years | 2 years | First cousins | ETFDH:c.807C > T, p.Gln269His; missense | Homozygous | As part of Family screening; NBS was not performed |
| 7 | Vomiting; recurrent viral illness; anemia | Female | 7 years | 8 days | First cousins | ETFDH:c.807C > T, p.Gln269His; missense | Homozygous | During NBS confirmatory workup |
| 8 | Mild glutaric aciduria type 2; Repaired double aortic arch | Male | 5 months | 5 months | First cousins | ETFDH:c.807C > T, p.Gln269His; missense | Homozygous | During NBS confirmatory workup |
| 9 | Asymptomatic GA-II; Respiratory problems | Female | 2 weeks | 2 weeks | Not related | ETFDH:c.807C > T, p.Gln269His; missense | Homozygous | During NBS confirmatory workup |
| 10 | Myopathy; Hypoglycemia; Recurrent vomiting; Nausea; Hepatitis | Male | 30 years | 30 years | NA | ETFDH:c.807C > T, p.Gln269His; missense | Homozygous | Based on clinical presentation; NBS was not performed |
| 11 | Encephalopathy; vomiting; hypoglycemia; metabolic acidosis | Female | 3 months | 4 months | NA | ETFDH:c.1325C > T p.Ser442Leu; missense | Homozygous | Based on clinical presentation; NBS was not performed |
| 12 | Metabolic crises; hypoglycemia; sepsis; hyperammonemia | Female | 2 days | 2 months | NA | ETFDH:c.1414G > A p.Gly472Arg; missense ETFB:c.278dup p.Pro94Thrfs*; Frameshift | Homozygous Heterozygous | Based on clinical presentation; NBS was not performed |
| 13 | Recurrent vomiting; Hypoglycemia; Seizures | Female | 4 years | 7 years | Not related | ETFB:c.1035del p.Ile346Phefs*19; frameshift | Homozygous | Based on clinical presentation; NBS was not performed |
| Case I.D | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acylcarnitine profile | Acetylcarnitine, C2 | NA | 4.03 RR = 2–17.83 | np | np | 31.9, H RR = 2–27.57 | np | 11.14 RR = 2–27.57 | 15.51 RR = 2.14–15.89 | 5.5 RR = 2–27.57 | 5.5 RR = 2.–17.8 | np | 1.46 RR = 2–15.89 | 2.18 RR = 2–27.57 |
| Iso-/Butyrylcarnitine, C4 | NA | 0.18 RR < 0.83 | np | np | 0.62 RR < 1.06 | np | 0.58, H RR < 1.06 | 0.64, H RR < 0.46 | 0.48 RR < 1.06 | 0.36 RR < 0.83 | np | 2.88, H RR < 0.46 | 0.78, H RR < 1.06 | |
| Isovaleryl-/2-Methylbutyrylcarn, C5 | NA | 0.07 RR < 0.51 | np | np | 0.98, H RR < 0.63 | np | 0.39 RR < 0.63 | 0.44, H RR < 0.38 | 1.04, H RR < 0.63 | 0.14 RR < 0.51 | np | 3.41, H RR < 0.38 | 0.49 RR < 0.63 | |
| Hexanoylcarnitine, C6 | NA | 0.08 RR < 0.17 | np | np | 0.56, H RR < 0.23 | np | 0.35, H RR < 0.23 | 0.68, H RR < 0.14 | 0.68, H RR < 0.23 | 0.23, H RR < 0.17 | np | 0.03 RR < 0.14 | 0.56, H RR < 0.23 | |
| Octanoylcarnitine, C8 | NA | 0.19 RR < 0.78 | np | np | 1.21, H RR < 0.45 | np | 0.72, H RR < 0.45 | 1.07, H RR < 0.19 | 2.09, H RR < 0.45 | 1.99, H RR < 0.78 | np | 0.12 RR < 0.19 | 3.38, H RR < 0.45 | |
| Decenoylcarnitine, C10:1 | NA | 0.11 RR < 0.27 | np | np | 0.27 RR < 0.46 | np | 0.2 RR < 0.46 | 0.39, H RR < 0.25 | 0.62, H RR < 0.46 | 0.39 RR < 0.47 | np | 0.03 RR < 0.25 | 0.94, H RR < 0.46 | |
| Decanoylcarnitine, C10 | NA | 0.21 RR < 0.88 | np | np | 1.46, H RR < 0.91 | np | 1.4, H RR < 0.91 | 1.93, H RR < 0.27 | 4, H RR < 0.91 | 2.79, H RR < 0.88 | np | 0.28, H RR < 0.27 | 5.77, H RR < 0.91 | |
| Glutarylcarnitine, C5-DC | NA | 0.10 RR < 0.11 | np | np | 0.06 RR < 0.1 | np | 0.12, H RR < 0.1 | 0.22, H RR < 0.06 | 0.1 RR < 0.1 | 0.13, H RR < 0.11 | np | 0.47, H RR < 0.06 | 0.14, H RR < 0.1 | |
| Dodecanoylcarnitine, C12 | NA | 0.08 RR < 0.26 | np | np | 0.35 RR < 0.35 | np | 1.17, H RR < 0.35 | 2.21, H RR < 0.18 | 0.83, H RR < 0.35 | 0.58, H RR < 0.26 | np | 0.56, H RR < 0.18 | 2.12, H RR < 0.35 | |
| Tetradecadienoylcarnitine, C14:2 | NA | 0.03 RR < 0.18 | np | np | 0.09 RR < 0.13 | np | 0.11 RR < 0.13 | 0.5, H RR < 0.09 | 0.19, H RR < 0.13 | 0.12 RR < 0.18 | np | 0.07 RR < 0.09 | 0.38, H RR < 0.13 | |
| Tetradecenoylcarnitine, C14:1 | NA | 0.07 RR < 0.24 | np | np | 0.22 RR < 0.35 | np | 0.58, H RR < 0.35 | 1.8, H RR < 0.16 | 0.45, H RR < 0.35 | 0.27, H RR < 0.24 | np | 0.48, H RR < 0.16 | 0.94, H RR < 0.35 | |
| Tetradecanoylcarnitine, C14 | NA | 0.04 RR < 0.12 | np | np | 0.17, H RR < 0.15 | np | 0.61, H RR < 0.15 | 1.64, H RR < 0.11 | 0.31, H RR < 0.15 | 0.14, H RR < 0.12 | np | 0.95, H RR < 0.11 | 0.57, H RR < 0.15 | |
| Hexadecenoylcarnitine, C16:1 | NA | 0.06 RR < 0.10 | np | np | 0.08 RR < 0.21 | np | 0.34, H RR < 0.21 | 1.43, H RR < 0.15 | 0.18 RR < 0.21 | 0.07 RR < 0.10 | np | 0.82, H RR < 0.15 | 0.53, H RR < 0.21 | |
| Hexadecanoylcarnitine, C16 | NA | 0.10 RR < 0.23 | np | np | 0.17 RR < 0.52 | np | 0.63, H RR < 0.52 | 1.84, H RR < 0.36 | 0.22 RR < 0.52 | 0.12 RR < 0.23 | np | 2.76, H RR < 0.36 | 0.4 RR < 0.52 | |
| Octadecanoylcarnitine, C18 | NA | 0.05 RR < 0.14 | np | np | 0.1 RR < 0.12 | np | 0.08 RR < 0.12 | 0.73, H RR < 0.1 | 0.11 RR < 0.12 | 0.03 RR < 0.14 | np | 0.58, H RR < 0.1 | 0.13, H RR < 0.12 | |
| Urine organic acid | np | np | + | N | Np | N | N | + | np | N | + | + | + | |
| Carnitine profile | Total (nmol/mL) | 45 RR = 19–59 | 42 RR = 34–78 | 81, H RR = 43–65 | 20 RR = 34–77 | 86, H RR = 35–84 | 16 RR = 35–84 | 133, H RR = 35–84 | 81, H RR = 17–41 | 52, H RR = 17–41 | 74 RR = 34–78 | np | 18 RR = 17–41 | 35 RR = 28–83 |
| Free (nmol/mL) | 24 RR = 12–46 | 36 RR = 25–54 | 44 RR = 30–50 | 14 RR = 22–65 | 42 RR = 24–63 | 12 RR = 24–63 | 53 RR = 24–63 | 44, H RR = 10–21 | 38, H RR = 10–21 | 64, H RR = 25–54 | np | 4 RR = 10–21 | 18 RR = 22–66 | |
| Acylcarnitine (nmol/mL) | 21, H RR = 4–15 | 6 RR = 5–30 | 7 RR = 7–14 | 6 RR = 4–29 | 52, H RR = 4–26 | 4 RR = 4–26 | 95, H RR = 4–28 | 37, H RR = 3–24 | 14 RR = 3–24 | 10 RR = 5–30 | np | 14 RR = 3–24 | 17 RR = 3–32 | |
| AC/FC ratio | 0.9 RR = 0.1–0.7 | 0.2 RR = 0.1–0.8 | np | 0.4 RR = 0.1–0.9 | 1.5, H RR = 0.1–0.8 | 0.3 RR = 0.1–0.8 | 2.5 RR = 0.1–0.8 | 0.8 RR = 0.1–0.8 | 0.4 RR = 0.1–0.8 | 0.2 RR = 0.1–0.8 | np | 3.5, H RR = 0.2–1.4 | 0.4 RR = 0.1–0.9 | |
| Glucose levels | Glucose (mmol/L) | 4.8 RR = 3.9–6.1 | 2.9, L RR = 3.9–6.1 | 3.4, L RR = 3.9–6.1 | 3.9 RR = 3.9–6.1 | 3.6, L RR = 3.9–6.1 | 3.4, L RR = 3.9–6.1 | 5.2 RR = 3.9–6.1 | 5.3 RR = 3.9–6.1 | 6.5, H RR = 3.9–6.1 | 2.9, L RR = 3.9–6.1 | np | 3.4, L RR = 3.9–6.1 | 2.8, L RR = 3.9–6.1 |
| Ammonia levels | Ammonia (µmol/L) | Np | 52, H RR = 9–35 | 56, H RR = 9–35 | 158, H RR = 15–51 | Np | 30.6 RR = 16–60 | 128, H RR = 9–35 | 62, H RR = 9–28 | 97, H RR = 16–60 | 80, H RR = 9–35 | 500, H RR = 9–35 | 58, H RR = 9–35 | |
| Ketones in urine | Ketones | Np | Negative | +4 | +3 | +1 | np | np | Negative | +1 | Negative | np | np | +3 |
| Gene | Variant | SIFT | Polyphen-2 | LRT | Mutation Taster | Mutation Assessor | FATHMM | PROVEAN | MetaSVM | MetaLR | REVEL | CADD | DANN | Condel | JSD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Score | Score | Score | Score | Score | Score | Score | Score | Score | Score | Score | Score | Score | Score | ||
| ETFDH | R41L | 0D | 0.021B | 0.523D | 0.999D | 0.496L | 0.725D | 0.642D | 0.780T | 0.897D | 0.7255D | 22.5 | 0.992 | 0.554D | 0.735 |
| ETFDH | Q269H | 0D | 0.905D | 0.843D | 0.999D | 0.987H | 0.834D | 0.806D | 0.969D | 0.944D | 0.834D | 20.3 | 0.956 | 0.705D | 0.819 |
| ETFDH | S442L | 0D | 1.0D | 0.845D | 0.810D | 0.99D | 0.954D | 0.873D | 0.990D | 0.985D | 0.984D | 26.1 | 0.992 | 0.732D | 0.809 |
| ETFDH | G472R | 0D | 0.999D | 0.843D | 0.810D | 0.988H | 0.986D | 0.900D | 0.986D | 0.982D | 0.986D | 32 | 0.999 | 0.719D | 0.780 |
| ETFB | P94Tfs*8 | - | - | - | B | - | - | - | - | - | - | - | - | - | - |
| ETFB | I346Ffs*19 | - | - | - | B | - | - | - | - | - | - | - | - | - | - |
| Variant | Stability | RI (0–10) | ΔΔG (Kcal/mol) |
|---|---|---|---|
| ETFDH:p.Arg41Leu | Decreased | 4 | −0.35 |
| ETFDH:p.Gln269His | Decreased | 8 | −0.87 |
| ETFDH:p.Ser442Leu | Decreased | 9 | −3.0 |
| ETFDH:p.Gly472Arg | Decreased | 2 | −0.24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.; Almesmari, F.S.A.; Dhahouri, N.A.; Saleh Ali, A.M.; Aldhanhani, M.A.A.M.A.; Vijayan, R.; Al Tenaiji, A.; Al Shamsi, A.; Hertecant, J.; Al Jasmi, F. Clinical, Biochemical, and Genetic Heterogeneity in Glutaric Aciduria Type II Patients. Genes 2021, 12, 1334. https://doi.org/10.3390/genes12091334
Ali A, Almesmari FSA, Dhahouri NA, Saleh Ali AM, Aldhanhani MAAMA, Vijayan R, Al Tenaiji A, Al Shamsi A, Hertecant J, Al Jasmi F. Clinical, Biochemical, and Genetic Heterogeneity in Glutaric Aciduria Type II Patients. Genes. 2021; 12(9):1334. https://doi.org/10.3390/genes12091334
Chicago/Turabian StyleAli, Amanat, Fatmah Saeed Ali Almesmari, Nahid Al Dhahouri, Arwa Mohammad Saleh Ali, Mohammed Ahmed Ali Mohamed Ahmed Aldhanhani, Ranjit Vijayan, Amal Al Tenaiji, Aisha Al Shamsi, Jozef Hertecant, and Fatma Al Jasmi. 2021. "Clinical, Biochemical, and Genetic Heterogeneity in Glutaric Aciduria Type II Patients" Genes 12, no. 9: 1334. https://doi.org/10.3390/genes12091334
APA StyleAli, A., Almesmari, F. S. A., Dhahouri, N. A., Saleh Ali, A. M., Aldhanhani, M. A. A. M. A., Vijayan, R., Al Tenaiji, A., Al Shamsi, A., Hertecant, J., & Al Jasmi, F. (2021). Clinical, Biochemical, and Genetic Heterogeneity in Glutaric Aciduria Type II Patients. Genes, 12(9), 1334. https://doi.org/10.3390/genes12091334