
Comparative Mitogenomics and Phylogenetic Analyses of Pentatomoidea (Hemiptera: Heteroptera)

 ,
,  ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Mitochondrial Genome Sequencing and Assembly
2.3. Mitochondrial Genome Annotation and Sequence Analysis
2.4. Phylogenetic Analysis
3. Results and Discussion
3.1. General Features of Pentatomoid Mitogenomes
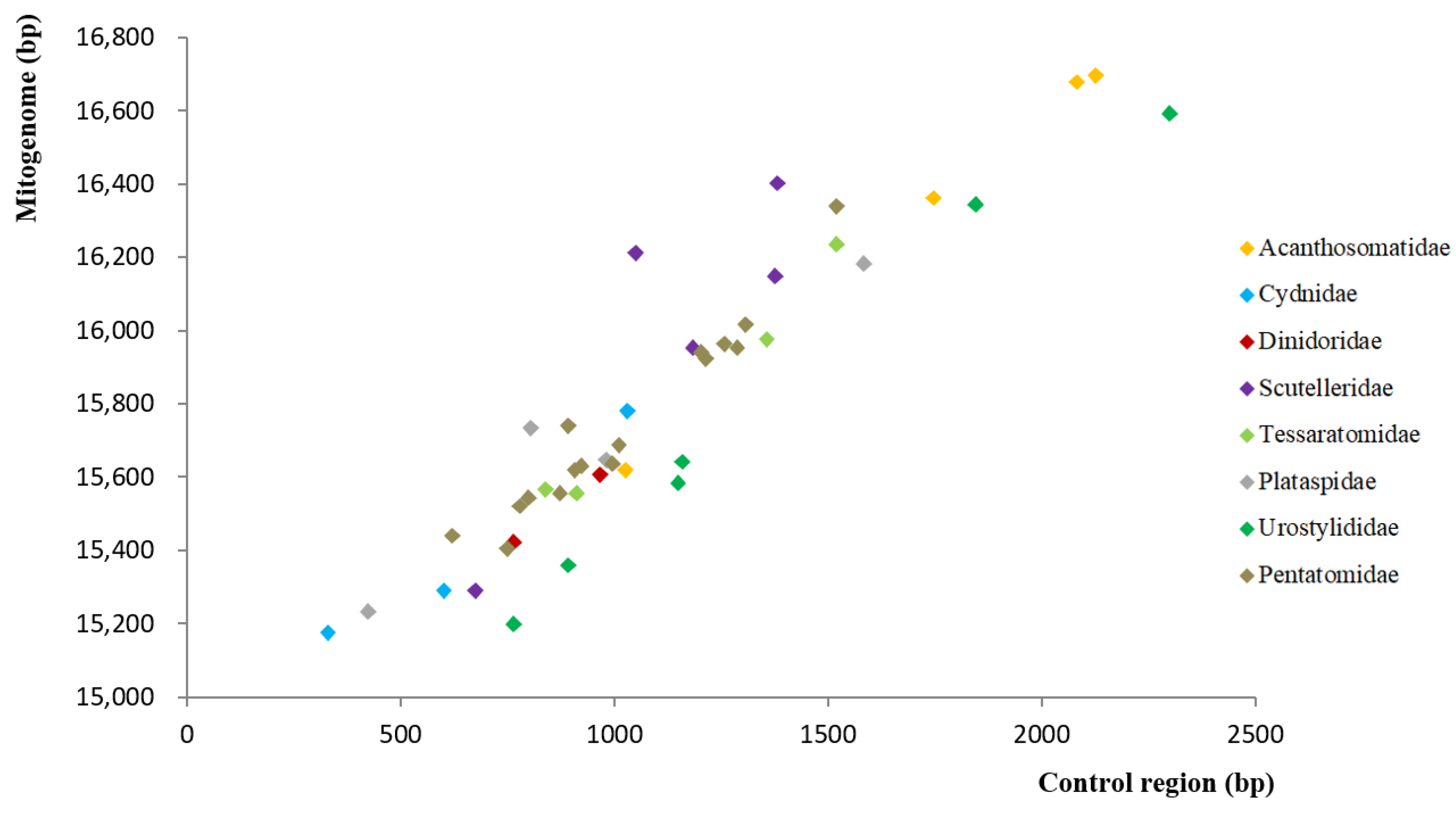
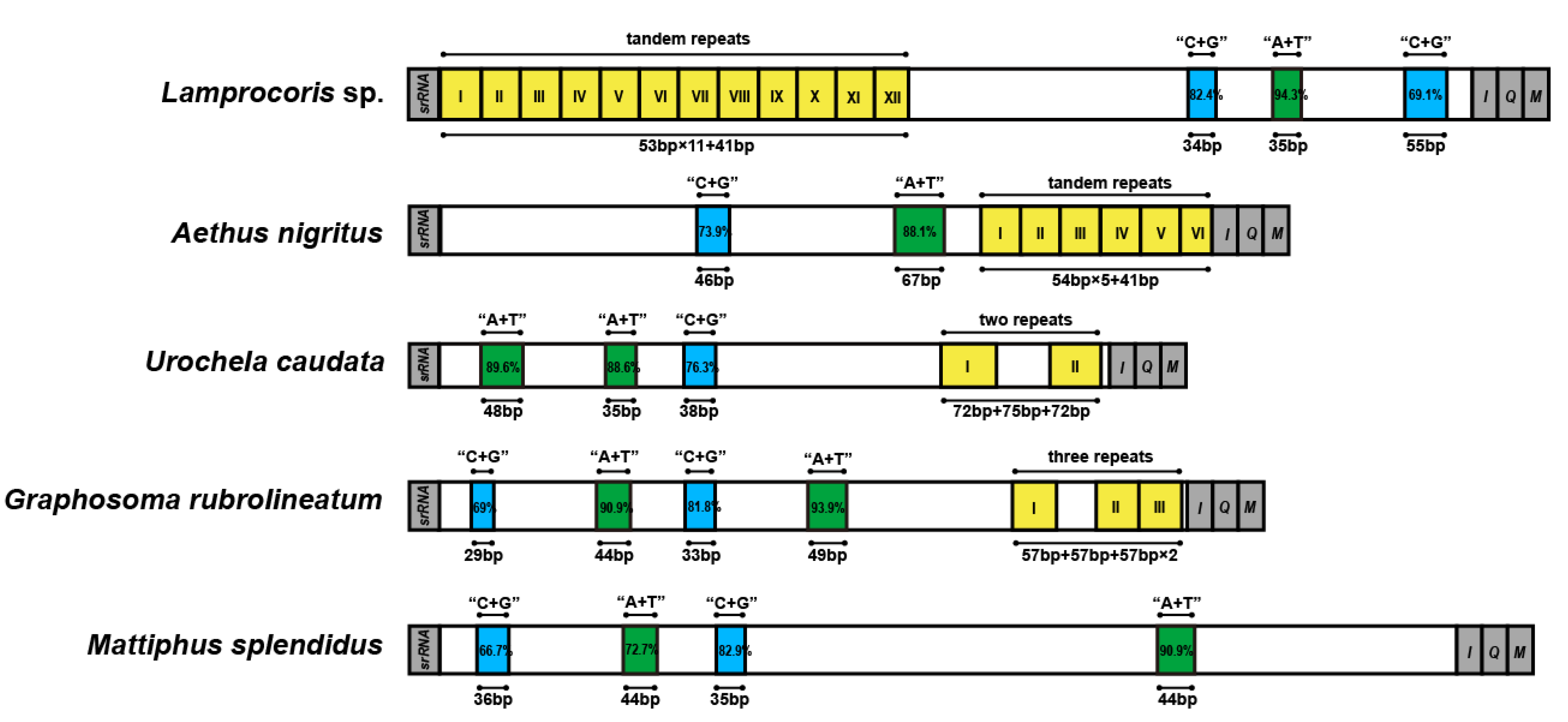
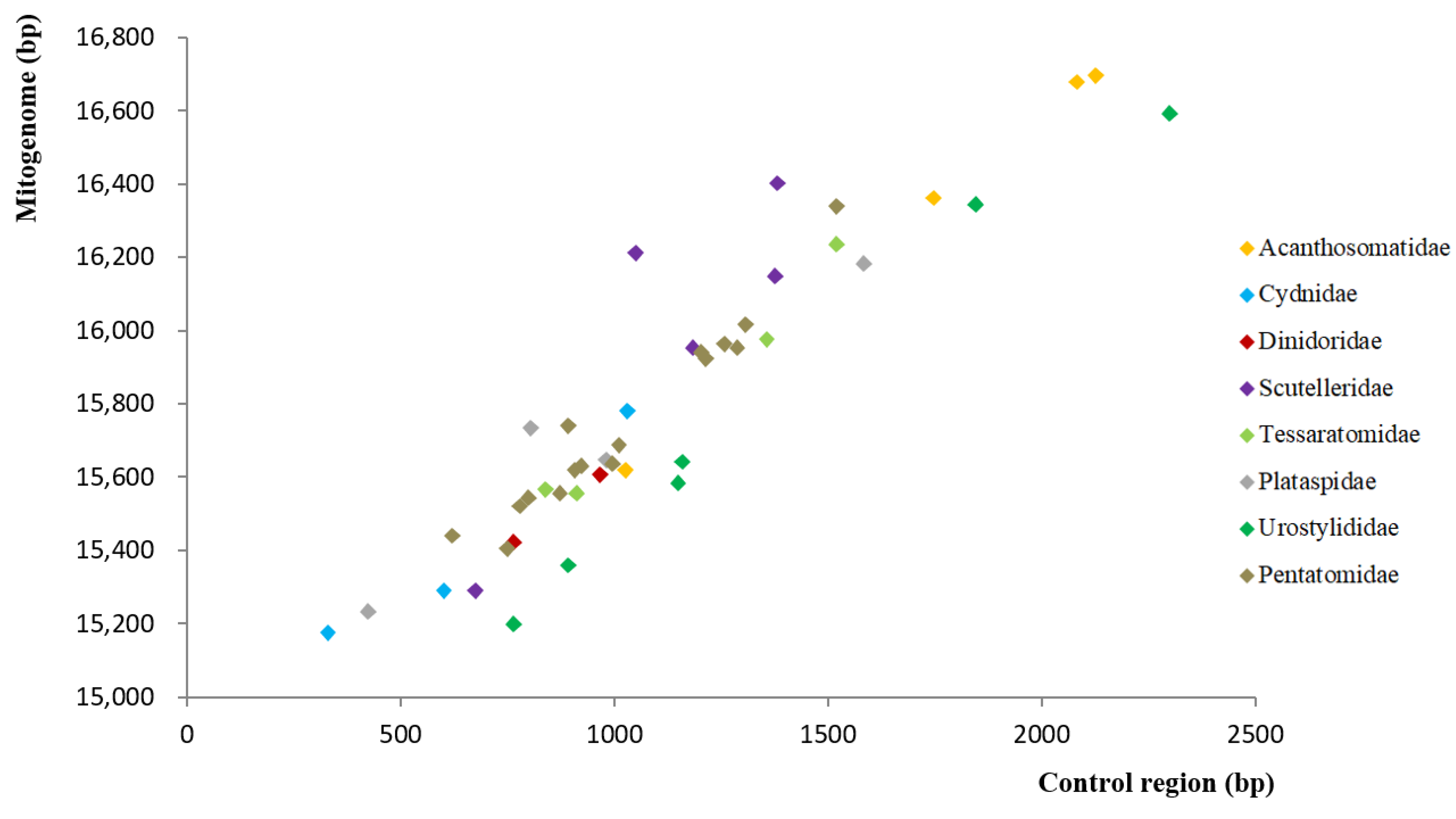
3.2. Non-Coding Regions (NCR) of Pentatomoid Mitogenomes
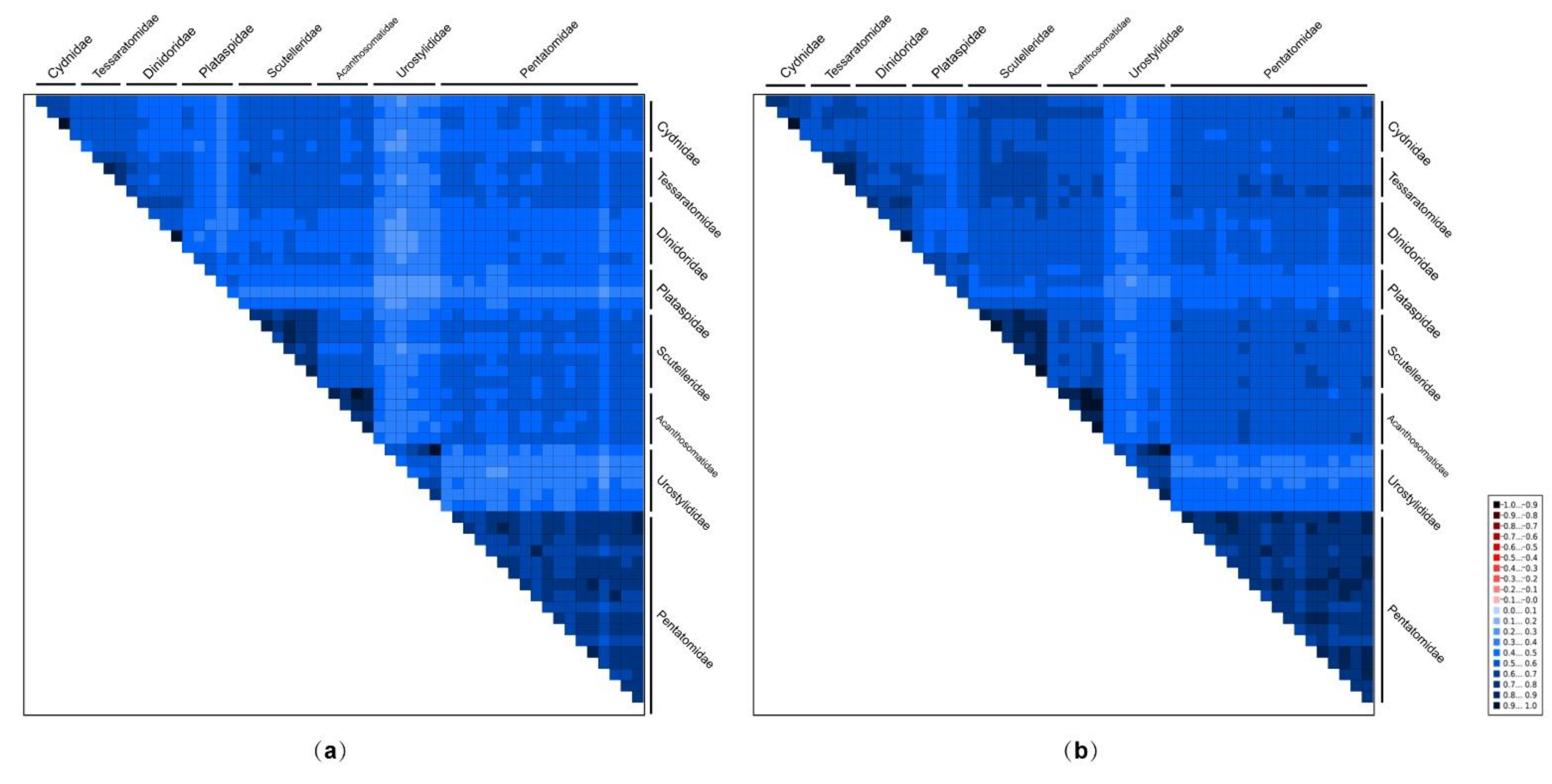
3.3. Sequence Heterogeneity in Pentatomoid Mitogenomes and Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McPherson, J.E. Invasive Stink Bugs and Related Species (Pentatomoidea); CRC Press: Boca Raton, FL, USA, 2018; p. 819. [Google Scholar]
- Rider, D.A. Pentatomoidea Home Page. North Dakota State University. Available online: https://www.ndsu.edu/faculty/rider/Pentatomoidea/ (accessed on 24 November 2015).
- Weirauch, C.; Schuh, R.T. Systematics and evolution of Heteroptera: 25 years of progress. Annu. Rev. Entomol. 2011, 56, 487–510. [Google Scholar] [CrossRef] [PubMed]
- Grazia, J.; Schuh, R.T.; Wheeler, W.C. Phylogenetic relationships of family groups in Pentatomoidea based on morphology and DNA sequences (Insecta: Heteroptera). Cladistics 2008, 24, 932–976. [Google Scholar] [CrossRef]
- Gapud, V. A generic revision of the subfamily Asopinae with consideration of its phylogenetic position in the family Pentatomidae and superfamily Pentatomoidea (Hemiptera-Heteroptera). Philipp. Entomol. 1991, 8, 865–961. [Google Scholar]
- Schuh, R.T.; Weirauch, C. True Bugs of the World (Hemiptera: Heteroptera) Classification and Natural History, 2nd ed.; Siri Scientific Press: Manchester, UK, 2020; p. 767. [Google Scholar]
- Yuan, M.L.; Zhang, Q.L.; Guo, Z.L.; Wang, J.; Shen, Y.Y. Comparative mitogenomic analysis of the superfamily Pentatomoidea (Insecta: Hemiptera: Heteroptera) and phylogenetic implications. BMC Genom. 2015, 16, 460. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.X.; Xie, Q.; Li, M.; Gao, C.Q.; Cui, Y.; Xi, L.; Bu, W.J. Phylogeny of pentatomomorphan bugs (Hemiptera-Heteroptera: Pentatomomorpha) based on six HOX gene fragments. Zootaxa 2011, 2888, 57–68. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Li, H.; Song, F.; Zhao, Y.S.; Wilson, J.J.; Cai, W.Z. Higher-level phylogeny and evolutionary history of Pentatomomorpha (Hemiptera: Heteroptera) inferred from mitochondrial genome sequences. Syst. Entomol. 2019, 44, 810–819. [Google Scholar] [CrossRef]
- Zhao, Q.; Wang, J.; Wang, M.Q.; Cai, B.; Wei, J.F. Complete mitochondrial genome of Dinorhynchus dybowskyi (Hemiptera: Pentatomidae: Asopinae) and phylogenetic analysis of Pentatomomorpha species. J. Insect Sci. 2018, 44, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.Q. Phylogeny and cladistic analysis of the Pentatomoidea (Hemiptera: Heteroptera). Acta Agric. Univ. Pekin. 1993, 19, 49–53. [Google Scholar]
- Lis, J.A.; Lis, P.; Ziaja, D.J.; Kocorek, A. Systematic position of Dinidoridae within the superfamily Pentatomoidea (Hemiptera: Heteroptera) revealed by the Bayesian phylogenetic analysis of the mitochondrial 12S and 16S rDNA sequences. Zootaxa 2012, 3423, 61–68. [Google Scholar] [CrossRef]
- Li, H.M.; Deng, R.Q.; Wang, J.W.; Chen, Z.Y.; Jia, F.L.; Wang, X.Z. A preliminary phylogeny of the Pentatomomorpha (Hemiptera: Heteroptera) based on nuclear 18S rDNA and mitochondrial DNA sequences. Mol. Phylogenet. Evol. 2005, 37, 313–326. [Google Scholar] [CrossRef]
- Wu, Y.Z.; Rédei, D.; Eger, J., Jr.; Wang, Y.H.; Wu, H.Y.; Carapezza, A.; Kment, P.; Cai, B.; Sun, X.Y.; Guo, P.L.; et al. Phylogeny and the colourful history of jewel bugs (Insecta: Hemiptera: Scutelleridae). Cladistics 2018, 34, 502–516. [Google Scholar] [CrossRef]
- Singh-Pruthi, H. The morphology of the male genitalia in Rhynchota. Trans. R. Entomol. Soc. Lond. 1925, 73, 127–267. [Google Scholar] [CrossRef]
- Li, H.; Shao, R.F.; Song, N.; Song, F.; Jiang, P.; Li, Z.H.; Cai, W.Z. Higher-level phylogeny of Paraneopteran insects inferred from mitochondrial genome sequences. Sci. Rep. 2015, 5, 8527. [Google Scholar] [CrossRef] [Green Version]
- Timmermans, M.J.T.N.; Barton, C.; Haran, J.; Ahrens, D.; Culverwell, C.L.; Ollikainen, A.; Dodsworth, S.; Foster, P.G.; Bocak, L.; Vogler, A.P. Family-level sampling of mitochondrial genomes in Coleoptera: Compositional heterogeneity and phylogenetics. Genome Biol. Evol. 2015, 8, 161–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, F.; Li, H.; Jiang, P.; Zhou, X.; Liu, J.; Sun, C.; Vogler, A.P.; Cai, W.Z. Capturing the phylogeny of Holometabola with mitochondrial genome data and Bayesian site-heterogeneous mixture models. Genome Biol. Evol. 2016, 8, 1411–1426. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Song, F.; Jiang, P.; Wilson, J.; Cai, W.Z.; Li, H. Compositional heterogeneity in true bug mitochondrial phylogenomics. Mol. Phylogenet. Evol. 2018, 118, 135–144. [Google Scholar] [CrossRef]
- Li, H.; Leavengood, J.M.; Chapman, E.G.; Burkhardt, D.; Song, F.; Jiang, P.; Liu, J.P.; Cai, W.Z. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. R. Soc. B 2017, 284, 20171223. [Google Scholar] [CrossRef]
- Nie, R.E.; Breeschoten, T.; Timmermans, M.J.T.N.; Nadein, K.; Xue, H.J.; Bai, M.; Huang, Y.; Yang, X.K.; Vogler, A.P. The phylogeny of Galerucinae (Coleoptera: Chrysomelidae) and the performance of mitochondrial genomes in phylogenetic inference compared to nuclear rRNA genes. Cladistics 2017, 34, 113–130. [Google Scholar] [CrossRef] [Green Version]
- Gillett, C.P.D.T.; Crampton-Platt, A.; Timmermans, M.J.T.N.; Jordal, B.H.; Emerson, B.C.; Vogler, A.P. Bulk de novo mitogenome assembly from pooled total DNA elucidates the phylogeny of weevils (Coleoptera: Curculionoidea). Mol. Biol. Evol. 2014, 31, 2223–2237. [Google Scholar] [CrossRef] [Green Version]
- Timmermans, M.J.T.N.; Lees, D.C.; Thompson, M.J.; Sáfián, S.; Brattström, O. Mitogenomics of “Old World Acraea” butterflies reveals a highly divergent “Bematistes”. Mol. Phylogenet. Evol. 2016, 97, 233–241. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Drummond, Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Meng, G.L.; Li, Y.Y.; Yang, C.T.; Liu, S.L. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kück, P.; Meid, S.A.; Groß, C.; Wägele, J.W.; Misof, B. AliGROOVE-visualization of heterogeneous sequence divergence within multiple sequence alignments and detection of inflated branch support. BMC Bioinform. 2014, 15, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanfear, R.; Calcott, B.; Ho, S.Y.W.; Guindon, S. PartitionFinder: Combined Selection of Partitioning Schemes and Substitution Models for Phylogenetic Analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [Green Version]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast-online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lartillot, N.; Rodrigue, N.; Stubbs, D.; Richer, J. PhyloBayes MPI: Phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 2013, 62, 611–615. [Google Scholar] [CrossRef] [Green Version]
- Lartillot, N.; Philippe, H.A. Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 2004, 21, 1095–1109. [Google Scholar] [CrossRef]
- Tavare, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
- Sun, Z.Q.; Liu, Y.Q.; Wilson, J.J.; Chen, Z.; Song, F.; Cai, W.Z.; Li, H. Mitochondrial genome of Phalantus geniculatus (Hemiptera: Reduviidae): trnT duplication and phylogenetic implications. Int. J. Biol. Macromol. 2019, 129, 110–115. [Google Scholar] [CrossRef]
- Zhao, Y.S.; Jiang, M.J.; Wu, Y.F.; Song, F.; Cai, W.Z.; Li, H. Mitochondrial genomes of three kissing bugs (Reduviidae: Triatominae) and their phylogenetic implications. Int. J. Biol. Macromol. 2019, 134, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Wu, Y.F.; Dai, R.H.; Yang, M.F. Comparative mitogenomes of six species in the subfamily Iassinae (Hemiptera: Cicadellidae) and phylogenetic analysis. Int. J. Biol. Macromol. 2020, 149, 1294–1303. [Google Scholar] [CrossRef]
- Yan, L.P.; Xu, W.T.; Zhang, D.; Li, J.Q. Comparative analysis of the mitochondrial genomes of flesh flies and their evolutionary implication. Int. J. Biol. Macromol. 2021, 174, 385–391. [Google Scholar] [CrossRef]
- Yua, L.L.; Ge, X.Y.; Xie, G.L.; Liu, H.Y.; Yang, Y.X. First Complete mitochondrial genome of Melyridae (Coleoptera, Cleroidea): Genome description and phylogenetic implications. Insects 2021, 12, 87. [Google Scholar]
- Wang, J.J.; Yang, M.F.; Dai, R.H.; Li, H.; Wang, X.Y. Characterization and phylogenetic implications of the complete mitochondrial genome of Idiocerinae (Hemiptera: Cicadellidae). Int. J. Biol. Macromol. 2018, 120, 2366–2372. [Google Scholar] [CrossRef]
- Zhao, Q.; Chen, C.; Liu, J.; Wei, J.F. Characterization of the complete mitochondrial genome of Eysarcoris aeneus (Heteroptera: Pentatomidae), with its phylogenetic analysis. Mitochondrial DNA Part B 2019, 4, 2096–2097. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Superfamily | Family | Species | GenBank Accession Number |
|---|---|---|---|
| Aradoidea | Aradidae | Aneurus similis | NC_030360 |
| Aradus compar | NC_030362 | ||
| Pyrrhocoroidea | Largidae | Macrocheraia grandis | MK809515 |
| Pyrrhocoridae | Pyrrhocoris tibialis | KX355214 | |
| Pentatomoidea | Acanthosomatidae | Microdeuterus sp. a | MW847242 |
| Sastragala esakiia | MW847247 | ||
| Acanthosoma labiduroides | JQ743670 | ||
| Anaxandra taurina | NC_042801 | ||
| Sastragala edessoides | JQ743676 | ||
| Cydnidae | Aethus nigritusa | MW847231 | |
| Macroscytus subaeneusa | MW847241 | ||
| Cydnidae sp. | MH643815 | ||
| Macroscytus gibbulus | NC_012457 | ||
| Scoparipes salvazai | NC_042800 | ||
| Dinidoridae | Eumenotes sp. a | MW847237 | |
| Megymenum gracilicorne | NC_042810 | ||
| Cyclopelta parva | NC_037739 | ||
| Coridius chinensis | JQ739179 | ||
| Megymenum brevicorne | JQ739181 | ||
| Scutelleridae | Chrysocoris stoliia | MW847234 | |
| Poecilocoris druraeia | MW847246 | ||
| Cantao ocellatus | NC_042803 | ||
| Eurygaster testudinaria | NC_042808 | ||
| Eucorysses grandis | JQ743671 | ||
| Lamprocoris sp. | JQ743674 | ||
| Poecilocoris nepalensis | JQ743675 | ||
| Tessaratomidae | Dalcantha dilatataa | MW847235 | |
| Mattiphus splendidus | MN496304 | ||
| Eusthenes cupreus | NC_022449 | ||
| Tessaratoma papillosa | NC_037742 | ||
| Plataspidae | Brachyplatys subaeneusa | MW847232 | |
| Calacta lugubrisa | MW847233 | ||
| Aponsila sp. | MF497710 | ||
| Coptosoma bifaria | NC_012449 | ||
| Megacopta cribraria | NC_015342 | ||
| Urostylididae | Urochela caudataa | MW847248 | |
| Urolabida histrionicaa | MW847249 | ||
| Urochela quadrinotata | NC_020144 | ||
| Urolabida sp. | MF497734 | ||
| Urostylis flavoannulata | NC_037747 | ||
| Urostylis sp. | JQ743679 | ||
| Pentatomidae | Dalpada cinctipesa | MW847236 | |
| Eurydema dominulusa | MW847238 | ||
| Glaucias dorsalisa | MW847239 | ||
| Hippotiscus dorsalisa | MW847240 | ||
| Neojurtina typicaa | MW847243 | ||
| Pentatoma metalliferaa | MW847244 | ||
| Plautia lushanicaa | MW847245 | ||
| Zicrona caeruleaa | MW847250 | ||
| Arma custos | MT535604 | ||
| Cazira horvathi | NC_042817 | ||
| Dalsira scabrata | NC_037374 | ||
| Dinorhynchus dybowskyi | NC_037724 | ||
| Eocanthecona thomsoni | NC_042816 | ||
| Gonopsis affinis | NC_036745 | ||
| Graphosoma rubrolineatum | NC_033875 | ||
| Pentatoma semiannulata | MT985377 | ||
| Picromerus griseus | NC_036418 | ||
| Scotinophara lurida | NC_042815 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, S.; Wu, Y.; Liu, Y.; Zhao, P.; Chen, Z.; Song, F.; Li, H.; Cai, W. Comparative Mitogenomics and Phylogenetic Analyses of Pentatomoidea (Hemiptera: Heteroptera). Genes 2021, 12, 1306. https://doi.org/10.3390/genes12091306
Xu S, Wu Y, Liu Y, Zhao P, Chen Z, Song F, Li H, Cai W. Comparative Mitogenomics and Phylogenetic Analyses of Pentatomoidea (Hemiptera: Heteroptera). Genes. 2021; 12(9):1306. https://doi.org/10.3390/genes12091306
Chicago/Turabian StyleXu, Shiwen, Yunfei Wu, Yingqi Liu, Ping Zhao, Zhuo Chen, Fan Song, Hu Li, and Wanzhi Cai. 2021. "Comparative Mitogenomics and Phylogenetic Analyses of Pentatomoidea (Hemiptera: Heteroptera)" Genes 12, no. 9: 1306. https://doi.org/10.3390/genes12091306
APA StyleXu, S., Wu, Y., Liu, Y., Zhao, P., Chen, Z., Song, F., Li, H., & Cai, W. (2021). Comparative Mitogenomics and Phylogenetic Analyses of Pentatomoidea (Hemiptera: Heteroptera). Genes, 12(9), 1306. https://doi.org/10.3390/genes12091306