
Advances in Genetic and Molecular Understanding of Alzheimer’s Disease


Abstract
1. Introduction
2. There Is More to Alzheimer’s Disease Than Amyloid
2.1. The Amyloid vs. Tau Hypotheses
2.2. Polyvalent APOE
2.3. The Underground of Alzheimer’s Disease—The Immune System
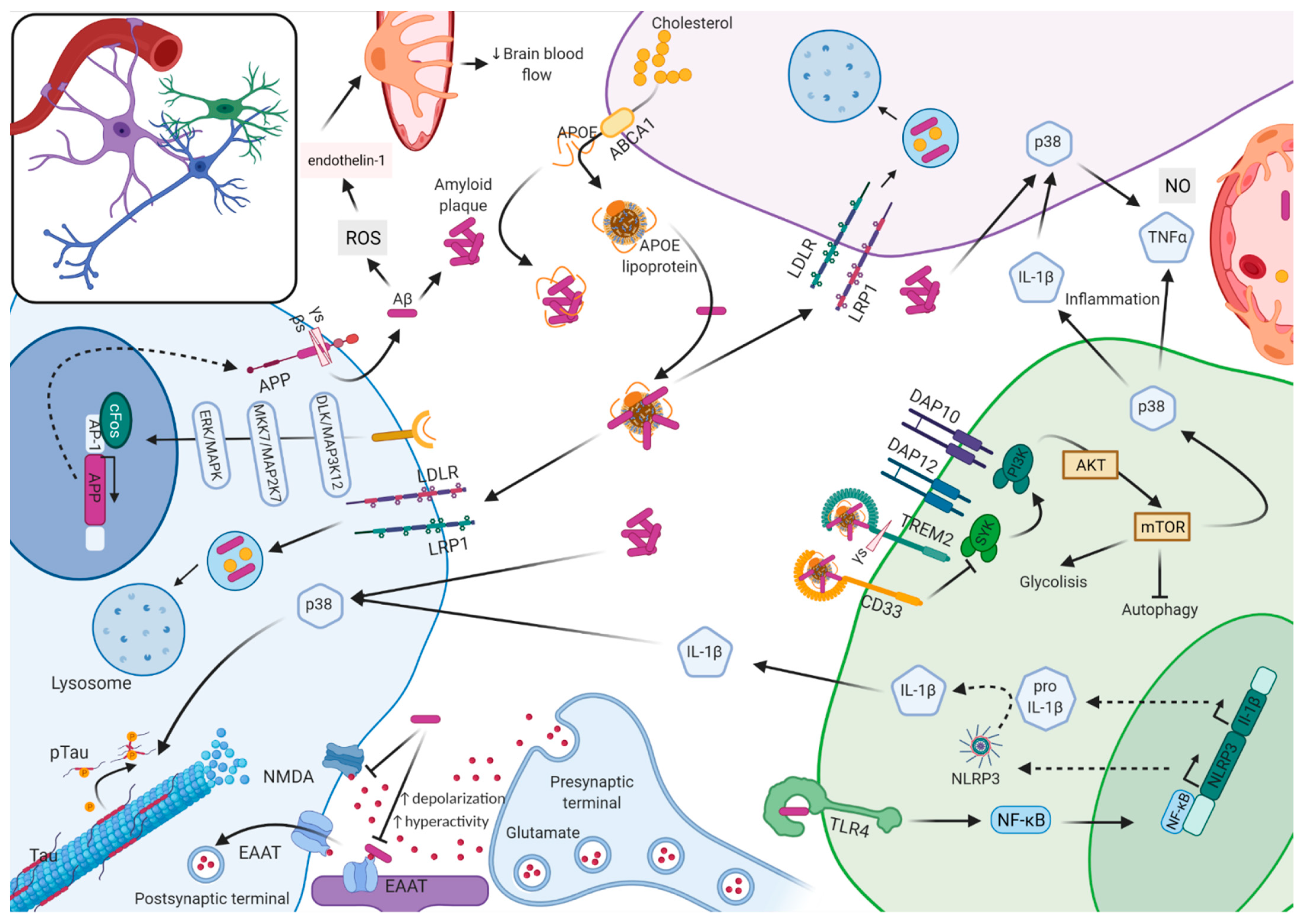
3. Unraveling the Molecular Mechanisms in AD Pathogenesis
4. Early Prediction and Diagnosis Are Key to Better Treatment
4.1. Risk Prediction and Prevention
4.2. Early Detection, Diagnosis, and Prognosis
4.3. Treatment
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5xFAD | Transgenic mouse model carrying APP KM670/671NL (Swedish), APP I716V (Florida), APP V717I (London), PSEN1 M146L (A>C), PSEN1 L286V mutations |
| AAO | Age at onset |
| ABCA7 | ATP-binding cassette subfamily A member 7 |
| ACE | Angiotensin I-converting enzyme |
| AD | Alzheimer’s disease |
| ADAD | Autosomal dominant Alzheimer’s disease |
| ADAM10 | ADAM metallopeptidase domain 10 |
| ALS | Amyotrohpic lateral sclerosis |
| ANK1 | Ankyrin 1 |
| APOE | Apolipoprotein E |
| APP | Amyloid precursor protein |
| APPNL-G-F | Knock-in mice model carrying the APP KM670/671NL (Swedish), APP I716F (Iberian), APP E693G (Arctic) mutations |
| APPswe/PS1dE9 | Transgenic mouse model carrying APP KM670/671NL (Swedish), PSEN1: deltaE9 mutations |
| APPPS1-21 | Transgenic mouse model carrying APP KM670/671NL (Swedish), PSEN1 L166P |
| ASO | Antisense oligonucleotides |
| Aβ | Amyloid β |
| BACE | β secretase, also known as β-site Amyloid precursor protein Ceaving Enzyme |
| BBB | Blood-brain barrier |
| BV2 | Immortalized murine microglial cell line |
| CD2AP | CD2-associated protein |
| CD33 | Myeloid cell surface antigen CD33 |
| CDR | Clinical dementia ratio |
| CE | Cholesterol ester |
| CERAD | Consortium to Establish a Registry for Alzheimer’s Disease |
| CELF1 | CUGBP Elav-like family member 1 |
| CLU | Clusterin |
| CNS | Central nervous system |
| CNV | Copy number variant |
| CR1 | Complement C3b/C4b receptor 1 (Knops blood group) |
| CSF | Cerebrospinal fluid |
| CT | Computerized tomography |
| DAM | Disease-associated microglia |
| DIAN | Dominantly inherited network |
| DLB | Dementia with Levy bodies |
| EMP | Erithro-myeloid progenitor |
| EAAT | Excitatory amino acid-mediated transporters |
| EOAD | Early onset Alzheimer’s disease |
| EPHA1 | EPH receptor A1 |
| ERK | Extracellular signal-regulated kinase |
| ETA | Endothelin A receptor |
| EWAs | Epigenome-wide association studies |
| fAD | Familial Alzheimer’s disease |
| fLOAD | Familial late-onset Alzheimer’s disease |
| FDR | False discovery rate |
| FTD | Frontotemporal dementia |
| GWA | Genome-wide association |
| H3K9ac | Histone 3 lysine 9 acetylation |
| H4K16ac | Histone 4 lysine 16 acetylation |
| HD | Huntington’s disease |
| HSPG | Heparan sulfate proteoglycan |
| iPSC | Induced pluripotent stem cells |
| ITAM | Immunoreceptor tyrosine-based activation motif |
| LDLR | Low-density lipoprotein receptor |
| LOAD | Late-onset Alzheimer’s disease |
| LRP1 | LDL (low-density lipoprotein) receptor-related protein 1 |
| MAP | Memory and aging project |
| MAPK | Mitogen-activated protein kinases (formerly known as ERK) |
| MAPT | Microtubule-associated protein tau |
| MCI | Mild cognitive impairment |
| MDMi | Monocyte-derived microglia-like cells |
| MKK7 | MAPK kinase 7 |
| MMSE | Mini-mental state examination |
| MOca | Montreal Cognitive Assessment |
| MRI | Magnetic resonance imaging |
| MS4A | Membrane spanning 4 |
| MS4A4A | Membrane spanning 4-domains A4A |
| mTOR | Mechanistic target of rapamycin |
| NfL | Neurofilament Ligtht |
| NFTs | Neuro fibrillary tangles |
| NGRN | Neurogranin |
| NLRP3 | Nucleotide-binding domain (NOD)-like receptor protein 3 |
| NOX4 | NADPH oxidase 4 |
| NT | Neuropil threads |
| p38MAPK | p38 mitogen-activated protein kinase |
| PD | Parkinson’s disease |
| PET | Positron emission tomography |
| PGRN | Progranulin |
| PI3K | Phosphatidylinositol-3-kinase |
| PIB | Pittsburgh compound B |
| PICALM | Phosphatidylinositol-binding clathrin assembly protein |
| PPAR | Peroxisome proliferator-activated receptor |
| PRS | Polygenic Risk Score |
| PS19 | Transgenic mouse model carrying MAPT P301S mutation |
| PSEN1 | Presenilin 1 |
| PSEN2 | Presenilin 2 |
| ROS | Religious Order Study |
| sAD | Sporadic Alzheimer’s disease |
| sLOAD | Sporadic late-onset Alzheimer’s disease |
| SNPs | Single nucleotide polymorphism |
| sPDGFRβ | Soluble platelet-derived growth factor receptor-β |
| SPI1 | Spi-1 proto-oncogene |
| TLRs | Toll-like receptors |
| TMEM106B | Transmembrane protein 106B |
| TREM2 | Triggering receptor expressed on myeloid cells 2 |
| TWAs | Transcriptome-wide association studies |
| TYROBP | Transmembrane immune signaling adaptor TYROBP |
| VD | Vascular dementia |
| VILIP1 | Visinin-like 1 |
References
- Livingston, G.; Sommerlad, A.; Orgeta, V.; Costafreda, S.G.; Huntley, J.; Ames, D.; Ballard, C.; Banerjee, S.; Burns, A.; Cohen-Mansfield, J.; et al. Dementia prevention, intervention, and care. Lancet 2017, 390, 2673–2734. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2019 Alzheimer’s Disease Facts and Figures. In Alzheimer’s & Dementia; Alzheimer’s Association: Chicago, IL, USA, 2019; p. 87. [Google Scholar]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Sims, R.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 2017, 49, 1373–1384. [Google Scholar] [CrossRef]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hagg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, J.D.; Ulland, T.K.; Mahan, T.E.; Nystrom, S.; Nilsson, K.P.; Song, W.M.; Zhou, Y.; Reinartz, M.; Choi, S.; Jiang, H.; et al. ApoE facilitates the microglial response to amyloid plaque pathology. J. Exp. Med. 2018, 215, 1047–1058. [Google Scholar] [CrossRef]
- Novikova, G.; Kapoor, M.; Tcw, J.; Abud, E.M.; Efthymiou, A.G.; Chen, S.X.; Cheng, H.; Fullard, J.F.; Bendl, J.; Liu, Y.; et al. Integration of Alzheimer’s disease genetics and myeloid genomics identifies disease risk regulatory elements and genes. Nat. Commun. 2021, 12, 1610. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, I.; Mauleon, A.; Rosense-Roca, M.; Alegret, M.; Vinyes, G.; Espinosa, A.; Sotolongo-Grau, O.; Becker, J.T.; Valero, S.; Tarraga, L.; et al. Identification of misdiagnosed fronto-temporal dementia using APOE genotype and phenotype-genotype correlation analyses. Curr. Alzheimer Res. 2014, 11, 182–191. [Google Scholar] [CrossRef][Green Version]
- Landau, S.M.; Horng, A.; Fero, A.; Jagust, W.J. Amyloid negativity in patients with clinically diagnosed Alzheimer disease and MCI. Neurology 2016, 86, 1377–1385. [Google Scholar] [CrossRef]
- van Dyck, C.H. Anti-Amyloid-β Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef]
- Cummings, J.; Ritter, A.; Zhong, K. Clinical Trials for Disease-Modifying Therapies in Alzheimer’s Disease: A Primer, Lessons Learned, and a Blueprint for the Future. J. Alzheimers Dis. 2018, 64 (Suppl. S1), S3–S22. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr1. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Gomez-Isla, T.; Hollister, R.; West, H.; Mui, S.; Growdon, J.H.; Petersen, R.C.; Parisi, J.E.; Hyman, B.T. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 1997, 41, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Isla, T.; Hollister, R.; West, H.; Mui, S.; Growdon, J.H.; Petersen, R.C.; Parisi, J.E.; Hyman, B.T. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch. Neurol. 2004, 61, 378–384. [Google Scholar]
- Bejanin, A.; Schonhaut, D.R.; La Joie, R.; Kramer, J.H.; Baker, S.L.; Sosa, N.; Ayakta, N.; Cantwell, A.; Janabi, M.; Lauriola, M.; et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 2017, 140, 3286–3300. [Google Scholar] [CrossRef]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365, eaav9518. [Google Scholar] [CrossRef]
- Zott, B.; Simon, M.M.; Hong, W.; Unger, F.; Chen-Engerer, H.J.; Frosch, M.P.; Sakmann, B.; Walsh, D.M.; Konnerth, A. A vicious cycle of β amyloid-dependent neuronal hyperactivation. Science 2019, 365, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- He, Z.; Guo, J.L.; McBride, J.D.; Narasimhan, S.; Kim, H.; Changolkar, L.; Zhang, B.; Gathagan, R.J.; Yue, C.; Dengler, C.; et al. Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 2018, 24, 29–38. [Google Scholar] [CrossRef]
- Maphis, N.; Jiang, S.; Xu, G.; Kokiko-Cochran, O.N.; Roy, S.M.; Van Eldik, L.J.; Watterson, D.M.; Lamb, B.T.; Bhaskar, K. Selective suppression of the α isoform of p38 MAPK rescues late-stage tau pathology. Alzheimers Res. Ther. 2016, 8, 54. [Google Scholar] [CrossRef]
- Ittner, A.; Chua, S.W.; Bertz, J.; Volkerling, A.; van der Hoven, J.; Gladbach, A.; Przybyla, M.; Bi, M.; van Hummel, A.; Stevens, C.H.; et al. Site-specific phosphorylation of tau inhibits amyloid-β toxicity in Alzheimer’s mice. Science 2016, 354, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Mass, E.; Jacome-Galarza, C.E.; Blank, T.; Lazarov, T.; Durham, B.H.; Ozkaya, N.; Pastore, A.; Schwabenland, M.; Chung, Y.R.; Rosenblum, M.K.; et al. A somatic mutation in erythro-myeloid progenitors causes neurodegenerative disease. Nature 2017, 549, 389–393. [Google Scholar] [CrossRef]
- Klein, H.U.; McCabe, C.; Gjoneska, E.; Sullivan, S.E.; Kaskow, B.J.; Tang, A.; Smith, R.V.; Xu, J.; Pfenning, A.R.; Bernstein, B.E.; et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat. Neurosci. 2019, 22, 37–46. [Google Scholar] [CrossRef]
- Getz, G.S.; Reardon, C.A. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J. Lipid Res. 2009, 50, S156–S161. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Frieden, C.; Garai, K. Structural differences between apoE3 and apoE4 may be useful in developing therapeutic agents for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2012, 109, 8913–8918. [Google Scholar] [CrossRef] [PubMed]
- Zhong, N.; Weisgraber, K.H. Understanding the association of apolipoprotein E4 with Alzheimer disease: Clues from its structure. J. Biol. Chem. 2009, 284, 6027–6031. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Herz, J.; Bu, G. Apolipoprotein E and apolipoprotein E receptors: Normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006312. [Google Scholar] [CrossRef]
- Mahley, R.W.; Rall, S.C., Jr. Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genom. Hum. Genet. 2000, 1, 507–537. [Google Scholar] [CrossRef]
- Mahley, R.W.; Rall, S.C., Jr. Apolipoprotein E; Apolipoprotein E genotype and cardiovascular disease in the Framingham Heart Study. Atherosclerosis 2001, 154, 529–537. [Google Scholar]
- Slooter, A.J.; Cruts, M.; Kalmijn, S.; Hofman, A.; Breteler, M.M.; Van Broeckhoven, C.; van Duijn, C.M. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: The Rotterdam Study. Arch. Neurol. 1998, 55, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Maestre, G.; Ottman, R.; Stern, Y.; Gurland, B.; Chun, M.; Tang, M.X.; Shelanski, M.; Tycko, B.; Mayeux, R. Apolipoprotein E and Alzheimer’s disease: Ethnic variation in genotypic risks. Ann. Neurol. 1995, 37, 254–259. [Google Scholar] [CrossRef] [PubMed]
- van der Lee, S.J.; Wolters, F.J.; Ikram, M.K.; Hofman, A.; Ikram, M.A.; Amin, N.; van Duijn, C.M. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: A community-based cohort study. Lancet Neurol. 2018, 17, 434–444. [Google Scholar] [CrossRef]
- Pastor, P.; Roe, C.M.; Villegas, A.; Bedoya, G.; Chakraverty, S.; Garcia, G.; Tirado, V.; Norton, J.; Rios, S.; Martinez, M.; et al. Apolipoprotein Eepsilon4 modifies Alzheimer’s disease onset in an E280A PS1 kindred. Ann. Neurol. 2003, 54, 163–169. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Risch, N.J.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C., Jr.; Rimmler, J.B.; Locke, P.A.; Conneally, P.M.; Schmader, K.E.; et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 1994, 7, 180–184. [Google Scholar] [CrossRef]
- Arboleda-Velasquez, J.F.; Lopera, F.; O’Hare, M.; Delgado-Tirado, S.; Marino, C.; Chmielewska, N.; Saez-Torres, K.L.; Amarnani, D.; Schultz, A.P.; Sperling, R.A.; et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: A case report. Nat. Med. 2019, 25, 1680–1683. [Google Scholar] [CrossRef]
- Hashimoto, T.; Serrano-Pozo, A.; Hori, Y.; Adams, K.W.; Takeda, S.; Banerji, A.O.; Mitani, A.; Joyner, D.; Thyssen, D.H.; Bacskai, B.J.; et al. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J. Neurosci. 2012, 32, 15181–15192. [Google Scholar] [CrossRef] [PubMed]
- Koffie, R.M.; Hashimoto, T.; Tai, H.C.; Kay, K.R.; Serrano-Pozo, A.; Joyner, D.; Hou, S.; Kopeikina, K.J.; Frosch, M.P.; Lee, V.M.; et al. Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-β. Brain 2012, 135, 2155–2168. [Google Scholar] [CrossRef]
- Christensen, D.Z.; Schneider-Axmann, T.; Lucassen, P.J.; Bayer, T.A.; Wirths, O. Accumulation of intraneuronal Abeta correlates with ApoE4 genotype. Acta Neuropathol. 2010, 119, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Zhang, J.; Liu, Q.; Liu, C.C.; Zhang, L.; Bu, G. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-β uptake. J. Neurosci. 2011, 31, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Castellano, J.M.; Jiang, H.; Basak, J.M.; Parsadanian, M.; Pham, V.; Mason, S.M.; Paul, S.M.; Holtzman, D.M. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular Aβ clearance. Neuron 2009, 64, 632–644. [Google Scholar] [CrossRef]
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef]
- Huang, Y.A.; Zhou, B.; Wernig, M.; Sudhof, T.C. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell 2017, 168, 427–441.e21. [Google Scholar] [CrossRef]
- Cruchaga, C.; Kauwe, J.S.; Harari, O.; Jin, S.C.; Cai, Y.; Karch, C.M.; Benitez, B.A.; Jeng, A.T.; Skorupa, T.; Carrell, D.; et al. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron 2013, 78, 256–268. [Google Scholar] [CrossRef]
- Kauwe, J.S.; Wang, J.; Mayo, K.; Morris, J.C.; Fagan, A.M.; Holtzman, D.M.; Goate, A.M. Alzheimer’s disease risk variants show association with cerebrospinal fluid amyloid β. Neurogenetics 2009, 10, 13–17. [Google Scholar] [CrossRef][Green Version]
- Deming, Y.; Li, Z.; Kapoor, M.; Harari, O.; Del-Aguila, J.L.; Black, K.; Carrell, D.; Cai, Y.; Fernandez, M.V.; Budde, J.; et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol. 2017, 133, 839–856. [Google Scholar] [CrossRef]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef]
- Pimenova, A.A.; Marcora, E.; Goate, A.M. A Tale of Two Genes: Microglial Apoe and Trem2. Immunity 2017, 47, 398–400. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef]
- Nugent, A.A.; Lin, K.; van Lengerich, B.; Lianoglou, S.; Przybyla, L.; Davis, S.S.; Llapashtica, C.; Wang, J.; Kim, D.J.; Xia, D.; et al. TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron 2020, 105, 837–854.e9. [Google Scholar] [CrossRef]
- Bertram, L.; Lange, C.; Mullin, K.; Parkinson, M.; Hsiao, M.; Hogan, M.F.; Schjeide, B.M.; Hooli, B.; Divito, J.; Ionita, I.; et al. Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 2008, 83, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Fitzpatrick, A.L.; Ikram, M.A.; DeStefano, A.L.; Gudnason, V.; Boada, M.; Bis, J.C.; Smith, A.V.; Carassquillo, M.M.; Lambert, J.C.; et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010, 303, 1832–1840. [Google Scholar] [CrossRef]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef]
- Benitez, B.A.; Jin, S.C.; Guerreiro, R.; Graham, R.; Lord, J.; Harold, D.; Sims, R.; Lambert, J.C.; Gibbs, J.R.; Bras, J.; et al. Missense variant in TREML2 protects against Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1510.e19. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.J.; Lohmann, E.; Bras, J.M.; Gibbs, J.R.; Rohrer, J.D.; Gurunlian, N.; Dursun, B.; Bilgic, B.; Hanagasi, H.; Gurvit, H.; et al. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 2013, 70, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.L.; Marcora, E.; Pimenova, A.A.; Di Narzo, A.F.; Kapoor, M.; Jin, S.C.; Harari, O.; Bertelsen, S.; Fairfax, B.P.; Czajkowski, J.; et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Manzine, P.R.; Ettcheto, M.; Cano, A.; Busquets, O.; Marcello, E.; Pelucchi, S.; Di Luca, M.; Endres, K.; Olloquequi, J.; Camins, A.; et al. ADAM10 in Alzheimer’s disease: Pharmacological modulation by natural compounds and its role as a peripheral marker. Biomed. Pharmacother. 2019, 113, 108661. [Google Scholar] [CrossRef]
- Jochemsen, H.M.; Teunissen, C.E.; Ashby, E.L.; van der Flier, W.M.; Jones, R.E.; Geerlings, M.I.; Scheltens, P.; Kehoe, P.G.; Muller, M. The association of angiotensin-converting enzyme with biomarkers for Alzheimer’s disease. Alzheimers Res. Ther. 2014, 6, 27. [Google Scholar] [CrossRef]
- Koronyo-Hamaoui, M.; Sheyn, J.; Hayden, E.Y.; Li, S.; Fuchs, D.T.; Regis, G.C.; Lopes, D.H.J.; Black, K.L.; Bernstein, K.E.; Teplow, D.B.; et al. Peripherally derived angiotensin converting enzyme-enhanced macrophages alleviate Alzheimer-related disease. Brain 2020, 143, 336–358. [Google Scholar] [CrossRef]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.I.; Mook-Jung, I.A. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e6. [Google Scholar] [CrossRef]
- Piers, T.M.; Cosker, K.; Mallach, A.; Johnson, G.T.; Guerreiro, R.; Hardy, J.; Pocock, J.M. A locked immunometabolic switch underlies TREM2 R47H loss of function in human iPSC-derived microglia. FASEB J. 2020, 34, 2436–2450. [Google Scholar] [CrossRef]
- Ulland, T.K.; Colonna, M. TREM2—A key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 667–675. [Google Scholar] [CrossRef]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid β. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Chowdhury, S.; Ma, R.; Le, K.X.; Hong, S.; Caldarone, B.J.; Stevens, B.; Lemere, C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017, 9, eaaf6295. [Google Scholar] [CrossRef] [PubMed]
- Ewers, M.; Franzmeier, N.; Suarez-Calvet, M.; Morenas-Rodriguez, E.; Caballero, M.A.A.; Kleinberger, G.; Piccio, L.; Cruchaga, C.; Deming, Y.; Dichgans, M.; et al. Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci. Transl. Med. 2019, 11, eaav6221. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Ulrich, J.D.; Finn, M.B.; Stewart, F.R.; Koscal, L.J.; Remolina Serrano, J.; Robinson, G.O.; Anderson, E.; Colonna, M.; Holtzman, D.M. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 11524–11529. [Google Scholar] [CrossRef] [PubMed]
- Leyns, C.E.G.; Gratuze, M.; Narasimhan, S.; Jain, N.; Koscal, L.J.; Jiang, H.; Manis, M.; Colonna, M.; Lee, V.M.Y.; Ulrich, J.D.; et al. TREM2 function impedes tau seeding in neuritic plaques. Nat. Neurosci. 2019, 22, 1217–1222. [Google Scholar] [CrossRef]
- Long, H.; Zhong, G.; Wang, C.; Zhang, J.; Zhang, Y.; Luo, J.; Shi, S. TREM2 Attenuates Abeta1-42-Mediated Neuroinflammation in BV-2 Cells by Downregulating TLR Signaling. Neurochem. Res. 2019, 44, 1830–1839. [Google Scholar] [CrossRef]
- Deming, Y.; Li, Z.; Benitez, B.A.; Cruchaga, C. Triggering receptor expressed on myeloid cells 2 (TREM2): A potential therapeutic target for Alzheimer disease? Expert Opin. Ther. Targets 2018, 22, 587–598. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef]
- Deming, Y.; Filipello, F.; Cignarella, F.; Cantoni, C.; Hsu, S.; Mikesell, R.; Li, Z.; Del-Aguila, J.L.; Dube, U.; Farias, F.G.; et al. The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk. Sci. Transl. Med. 2019, 11, eaau2291. [Google Scholar] [CrossRef]
- Kwart, D.; Gregg, A.; Scheckel, C.; Murphy, E.A.; Paquet, D.; Duffield, M.; Fak, J.; Olsen, O.; Darnell, R.B.; Tessier-Lavigne, M. A Large Panel of Isogenic APP and PSEN1 Mutant Human iPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP β-CTFs, Not Aβ. Neuron 2019, 104, 256–270.e5. [Google Scholar] [CrossRef]
- Van Acker, Z.P.; Bretou, M.; Annaert, W. Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: Impact of genetic risk factors. Mol. Neurodegener. 2019, 14, 20. [Google Scholar] [CrossRef]
- Kwart, D.; Gregg, A.; Scheckel, C.; Murphy, E.A.; Paquet, D.; Duffield, M.; Fak, J.; Olsen, O.; Darnell, R.B.; Tessier-Lavigne, M.A. LC3-Associated Endocytosis Facilitates β-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell 2019, 178, 536–551.e14. [Google Scholar]
- Jun, G.; Ibrahim-Verbaas, C.A.; Vronskaya, M.; Lambert, J.C.; Chung, J.; Naj, A.C.; Kunkle, B.W.; Wang, L.S.; Bis, J.C.; Bellenguez, C.; et al. A novel Alzheimer disease locus located near the gene encoding tau protein. Mol. Psychiatry 2016, 21, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Del-Aguila, J.L.; Dube, U.; Budde, J.; Martinez, R.; Black, K.; Xiao, Q.; Cairns, N.J.; Dominantly Inherited Alzheimer, N.; Dougherty, J.D.; et al. Genetic variants associated with Alzheimer’s disease confer different cerebral cortex cell-type population structure. Genome Med. 2018, 10, 43. [Google Scholar] [CrossRef]
- Li, Z.; Farias, F.H.G.; Dube, U.; Del-Aguila, J.L.; Mihindukulasuriya, K.A.; Fernandez, M.V.; Ibanez, L.; Budde, J.P.; Wang, F.; Lake, A.M.; et al. The TMEM106B FTLD-protective variant, rs1990621, is also associated with increased neuronal proportion. Acta Neuropathol. 2020, 139, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Lodato, M.A.; Rodin, R.E.; Bohrson, C.L.; Coulter, M.E.; Barton, A.R.; Kwon, M.; Sherman, M.A.; Vitzthum, C.M.; Luquette, L.J.; Yandava, C.N.; et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 2018, 359, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Verheijen, B.M.; Vermulst, M.; van Leeuwen, F.W. Somatic mutations in neurons during aging and neurodegeneration. Acta Neuropathol. 2018, 135, 811–826. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H. Somatic mutations in disorders with disrupted brain connectivity. Exp. Mol. Med. 2016, 48, e239. [Google Scholar] [CrossRef]
- Bushman, D.M.; Kaeser, G.E.; Siddoway, B.; Westra, J.W.; Rivera, R.R.; Rehen, S.K.; Yung, Y.C.; Chun, J. Genomic mosaicism with increased amyloid precursor protein (APP) gene copy number in single neurons from sporadic Alzheimer’s disease brains. eLife 2015, 4, e05116. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Acuna-Hidalgo, R.; Keogh, M.J.; Quenez, O.; Steehouwer, M.; Lelieveld, S.; Rousseau, S.; Richard, A.C.; Oud, M.S.; Marguet, F.; et al. Somatic variants in autosomal dominant genes are a rare cause of sporadic Alzheimer’s disease. Alzheimers Dement. 2018, 14, 1632–1639. [Google Scholar] [CrossRef]
- Annese, A.; Manzari, C.; Lionetti, C.; Picardi, E.; Horner, D.S.; Chiara, M.; Caratozzolo, M.F.; Tullo, A.; Fosso, B.; Pesole, G.; et al. Whole transcriptome profiling of Late-Onset Alzheimer’s Disease patients provides insights into the molecular changes involved in the disease. Sci. Rep. 2018, 8, 4282. [Google Scholar] [CrossRef]
- Li, X.; Long, J.; He, T.; Belshaw, R.; Scott, J. Integrated genomic approaches identify major pathways and upstream regulators in late onset Alzheimer’s disease. Sci. Rep. 2015, 5, 12393. [Google Scholar] [CrossRef]
- Wang, M.; Roussos, P.; McKenzie, A.; Zhou, X.; Kajiwara, Y.; Brennand, K.J.; De Luca, G.C.; Crary, J.F.; Casaccia, P.; Buxbaum, J.D.; et al. Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer’s disease. Genome Med. 2016, 8, 104. [Google Scholar] [CrossRef]
- Caberlotto, L.; Marchetti, L.; Lauria, M.; Scotti, M.; Parolo, S. Integration of transcriptomic and genomic data suggests candidate mechanisms for APOE4-mediated pathogenic action in Alzheimer’s disease. Sci. Rep. 2016, 6, 32583. [Google Scholar] [CrossRef]
- Mostafavi, S.; Gaiteri, C.; Sullivan, S.E.; White, C.C.; Tasaki, S.; Xu, J.; Taga, M.; Klein, H.U.; Patrick, E.; Komashko, V.; et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci. 2018, 21, 811–819. [Google Scholar] [CrossRef]
- Sepulcre, J.; Grothe, M.J.; d’Oleire Uquillas, F.; Ortiz-Teran, L.; Diez, I.; Yang, H.S.; Jacobs, H.I.L.; Hanseeuw, B.J.; Li, Q.; El-Fakhri, G.; et al. Neurogenetic contributions to amyloid β and tau spreading in the human cortex. Nat. Med. 2018, 24, 1910–1918. [Google Scholar] [CrossRef]
- Del-Aguila, J.L.; Li, Z.; Dube, U.; Mihindukulasuriya, K.A.; Budde, J.P.; Fernandez, M.V.; Ibanez, L.; Bradley, J.; Wang, F.; Bergmann, K.; et al. A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimers Res. Ther. 2019, 11, 71. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K.; Friedman, B.A.; Etxeberria, A.; Huntley, M.A.; van der Brug, M.P.; Foreman, O.; Paw, J.S.; Modrusan, Z.; Beach, T.G.; Serrano, G.E.; et al. Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep. 2020, 31, 107843. [Google Scholar] [CrossRef] [PubMed]
- Dube, U.; Del-Aguila, J.L.; Li, Z.; Budde, J.P.; Jiang, S.; Hsu, S.; Ibanez, L.; Fernandez, M.V.; Farias, F.; Norton, J.; et al. An atlas of cortical circular RNA expression in Alzheimer disease brains demonstrates clinical and pathological associations. Nat. Neurosci. 2019, 22, 1903–1912. [Google Scholar] [CrossRef]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Lunnon, K.; Smith, R.; Hannon, E.; De Jager, P.L.; Srivastava, G.; Volta, M.; Troakes, C.; Al-Sarraj, S.; Burrage, J.; Macdonald, R.; et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat. Neurosci. 2014, 17, 1164–1170. [Google Scholar] [CrossRef] [PubMed]
- Gasparoni, G.; Bultmann, S.; Lutsik, P.; Kraus, T.F.J.; Sordon, S.; Vlcek, J.; Dietinger, V.; Steinmaurer, M.; Haider, M.; Mulholland, C.B.; et al. DNA methylation analysis on purified neurons and glia dissects age and Alzheimer’s disease-specific changes in the human cortex. Epigenet. Chromatin 2018, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.R.; Smith, R.G.; Burrage, J.; Troakes, C.; Al-Sarraj, S.; Kalaria, R.N.; Sloan, C.; Robinson, A.C.; Mill, J.; Lunnon, K. A cross-brain regions study of ANK1 DNA methylation in different neurodegenerative diseases. Neurobiol. Aging 2019, 74, 70–76. [Google Scholar] [CrossRef]
- Nativio, R.; Donahue, G.; Berson, A.; Lan, Y.; Amlie-Wolf, A.; Tuzer, F.; Toledo, J.B.; Gosai, S.J.; Gregory, B.D.; Torres, C.; et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 497–505. [Google Scholar] [CrossRef]
- Pagani, M.; Nobili, F.; Morbelli, S.; Arnaldi, D.; Giuliani, A.; Oberg, J.; Girtler, N.; Brugnolo, A.; Picco, A.; Bauckneht, M.; et al. Early identification of MCI converting to AD: A FDG PET study. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 2042–2052. [Google Scholar] [CrossRef]
- Paglia, G.; Stocchero, M.; Cacciatore, S.; Lai, S.; Angel, P.; Alam, M.T.; Keller, M.; Ralser, M.; Astarita, G. Unbiased Metabolomic Investigation of Alzheimer’s Disease Brain Points to Dysregulation of Mitochondrial Aspartate Metabolism. J. Proteome Res. 2016, 15, 608–618. [Google Scholar] [CrossRef]
- Snowden, S.G.; Ebshiana, A.A.; Hye, A.; An, Y.; Pletnikova, O.; O’Brien, R.; Troncoso, J.; Legido-Quigley, C.; Thambisetty, M. Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: A nontargeted metabolomic study. PLoS Med. 2017, 14, e1002266. [Google Scholar] [CrossRef]
- Guiraud, S.P.; Montoliu, I.; Da Silva, L.; Dayon, L.; Galindo, A.N.; Corthesy, J.; Kussmann, M.; Martin, F.P. High-throughput and simultaneous quantitative analysis of homocysteine-methionine cycle metabolites and co-factors in blood plasma and cerebrospinal fluid by isotope dilution LC-MS/MS. Anal. Bioanal. Chem. 2017, 409, 295–305. [Google Scholar] [CrossRef]
- Toledo, J.B.; Arnold, M.; Kastenmuller, G.; Chang, R.; Baillie, R.A.; Han, X.; Thambisetty, M.; Tenenbaum, J.D.; Suhre, K.; Thompson, J.W.; et al. Metabolic network failures in Alzheimer’s disease: A biochemical road map. Alzheimers Dement. 2017, 13, 965–984. [Google Scholar] [CrossRef] [PubMed]
- Varma, V.R.; Oommen, A.M.; Varma, S.; Casanova, R.; An, Y.; Andrews, R.M.; O’Brien, R.; Pletnikova, O.; Troncoso, J.C.; Toledo, J.; et al. Brain and blood metabolite signatures of pathology and progression in Alzheimer disease: A targeted metabolomics study. PLoS Med. 2018, 15, e1002482. [Google Scholar] [CrossRef]
- Wilkins, J.M.; Trushina, E. Application of Metabolomics in Alzheimer’s Disease. Front. Neurol. 2017, 8, 719. [Google Scholar] [CrossRef]
- Yang, J.; Li, S.; He, X.B.; Cheng, C.; Le, W. Induced pluripotent stem cells in Alzheimer’s disease: Applications for disease modeling and cell-replacement therapy. Mol. Neurodegener. 2016, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.J.; White, C.C.; Patel, K.; Xu, J.; Olah, M.; Replogle, J.M.; Frangieh, M.; Cimpean, M.; Winn, P.; McHenry, A.; et al. A human microglia-like cellular model for assessing the effects of neurodegenerative disease gene variants. Sci. Transl. Med. 2017, 9, eaai7635. [Google Scholar] [CrossRef]
- Karch, C.M.; Hernandez, D.; Wang, J.C.; Marsh, J.; Hewitt, A.W.; Hsu, S.; Norton, J.; Levitch, D.; Donahue, T.; Sigurdson, W.; et al. Human fibroblast and stem cell resource from the Dominantly Inherited Alzheimer Network. Alzheimers Res. Ther. 2018, 10, 69. [Google Scholar] [CrossRef]
- Tcw, J. Human iPSC application in Alzheimer’s disease and Tau-related neurodegenerative diseases. Neurosci. Lett. 2019, 699, 31–40. [Google Scholar] [CrossRef]
- Xiang, X.; Piers, T.M.; Wefers, B.; Zhu, K.; Mallach, A.; Brunner, B.; Kleinberger, G.; Song, W.; Colonna, M.; Herms, J.; et al. The Trem2 R47H Alzheimer’s risk variant impairs splicing and reduces Trem2 mRNA and protein in mice but not in humans. Mol. Neurodegener. 2018, 13, 49. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wetzel, I.; Marriott, I.; Dreau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef]
- Matsui, T.K.; Tsuru, Y.; Hasegawa, K.; Kuwako, K.I. Vascularization of human brain organoids. Stem Cells 2021, 39, 1017–1024. [Google Scholar] [PubMed]
- Morris, J.C. The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology 1993, 43, 2412–2414. [Google Scholar] [CrossRef] [PubMed]
- Folstein, M.F.; Folstein, S.E.; Mchugh, P.R. Mini-Mental State—Practical Method for Grading Cognitive State of Patients for Clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Nasreddine, Z.S.; Phillips, N.A.; Bedirian, V.; Charbonneau, S.; Whitehead, V.; Collin, I.; Cummings, J.L.; Chertkow, H. The Montreal Cognitive Assessment, MoCA: A brief screening tool for mild cognitive impairment. J. Am. Geriatr. Soc. 2005, 53, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, H.C.; Munro Cullum, C.; Hynan, L.S.; Lacritz, L.H. The CERAD Neuropsychologic Battery Total Score and the progression of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2010, 24, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Cruchaga, C.; Del-Aguila, J.L.; Saef, B.; Black, K.; Fernandez, M.V.; Budde, J.; Ibanez, L.; Deming, Y.; Kapoor, M.; Tosto, G.; et al. Polygenic risk score of sporadic late-onset Alzheimer’s disease reveals a shared architecture with the familial and early-onset forms. Alzheimers Dement. 2018, 14, 205–214. [Google Scholar] [CrossRef]
- Darst, B.F.; Koscik, R.L.; Racine, A.M.; Oh, J.M.; Krause, R.A.; Carlsson, C.M.; Zetterberg, H.; Blennow, K.; Christian, B.T.; Bendlin, B.B.; et al. Pathway-Specific Polygenic Risk Scores as Predictors of Amyloid-β Deposition and Cognitive Function in a Sample at Increased Risk for Alzheimer’s Disease. J. Alzheimers Dis. 2017, 55, 473–484. [Google Scholar] [CrossRef]
- Kauppi, K.; Fan, C.C.; McEvoy, L.K.; Holland, D.; Tan, C.H.; Chen, C.H.; Andreassen, O.A.; Desikan, R.S.; Dale, A.M. Combining Polygenic Hazard Score with Volumetric MRI and Cognitive Measures Improves Prediction of Progression From Mild Cognitive Impairment to Alzheimer’s Disease. Front. Neurosci. 2018, 12, 260. [Google Scholar] [CrossRef]
- Tan, C.H.; Fan, C.C.; Mormino, E.C.; Sugrue, L.P.; Broce, I.J.; Hess, C.P.; Dillon, W.P.; Bonham, L.W.; Yokoyama, J.S.; Karch, C.M.; et al. Polygenic hazard score: An enrichment marker for Alzheimer’s associated amyloid and tau deposition. Acta Neuropathol. 2018, 135, 85–93. [Google Scholar] [CrossRef]
- Ferrari, R.; Wang, Y.; Vandrovcova, J.; Guelfi, S.; Witeolar, A.; Karch, C.M.; Schork, A.J.; Fan, C.C.; Brewer, J.B.; International, F.T.D.G.C.; et al. Genetic architecture of sporadic frontotemporal dementia and overlap with Alzheimer’s and Parkinson’s diseases. J. Neurol. Neurosurg. Psychiatry 2017, 88, 152–164. [Google Scholar] [CrossRef]
- Suarez-Calvet, M.; Araque Caballero, M.A.; Kleinberger, G.; Bateman, R.J.; Fagan, A.M.; Morris, J.C.; Levin, J.; Danek, A.; Ewers, M.; Haass, C.; et al. Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci. Transl. Med. 2016, 8, 369ra178. [Google Scholar] [CrossRef]
- Preische, O.; Schultz, S.A.; Apel, A.; Kuhle, J.; Kaeser, S.A.; Barro, C.; Graber, S.; Kuder-Buletta, E.; LaFougere, C.; Laske, C.; et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat. Med. 2019, 25, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Escott-Price, V.; Sims, R.; Bannister, C.; Harold, D.; Vronskaya, M.; Majounie, E.; Badarinarayan, N.; GERAD/PERADES; IGAP Consortia; Morgan, K.; et al. Common polygenic variation enhances risk prediction for Alzheimer’s disease. Brain 2015, 138, 3673–3684. [Google Scholar] [CrossRef] [PubMed]
- Escott-Price, V.; Shoai, M.; Pither, R.; Williams, J.; Hardy, J. Polygenic score prediction captures nearly all common genetic risk for Alzheimer’s disease. Neurobiol. Aging 2017, 49, 214.e7–214.e11. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Wiste, H.J.; Therneau, T.M.; Weigand, S.D.; Knopman, D.S.; Mielke, M.M.; Lowe, V.J.; Vemuri, P.; Machulda, M.M.; Schwarz, C.G.; et al. Associations of Amyloid, Tau, and Neurodegeneration Biomarker Profiles with Rates of Memory Decline Among Individuals Without Dementia. JAMA 2019, 321, 2316–2325. [Google Scholar] [CrossRef]
- Kester, M.I.; Teunissen, C.E.; Sutphen, C.; Herries, E.M.; Ladenson, J.H.; Xiong, C.; Scheltens, P.; van der Flier, W.M.; Morris, J.C.; Holtzman, D.M.; et al. Cerebrospinal fluid VILIP-1 and YKL-40, candidate biomarkers to diagnose, predict and monitor Alzheimer’s disease in a memory clinic cohort. Alzheimers Res. Ther. 2015, 7, 59. [Google Scholar] [CrossRef]
- Casaletto, K.B.; Elahi, F.M.; Bettcher, B.M.; Neuhaus, J.; Bendlin, B.B.; Asthana, S.; Johnson, S.C.; Yaffe, K.; Carlsson, C.; Blennow, K.; et al. Neurogranin, a synaptic protein, is associated with memory independent of Alzheimer biomarkers. Neurology 2017, 89, 1782–1788. [Google Scholar] [CrossRef]
- Nilselid, A.M.; Davidsson, P.; Nagga, K.; Andreasen, N.; Fredman, P.; Blennow, K. Clusterin; Clusterin in cerebrospinal fluid: Analysis of carbohydrates and quantification of native and glycosylated forms. Neurochem. Int. 2006, 48, 718–728. [Google Scholar] [CrossRef]
- Piccio, L.; Buonsanti, C.; Cella, M.; Tassi, I.; Schmidt, R.E.; Fenoglio, C.; Rinker, J., 2nd; Naismith, R.T.; Panina-Bordignon, P.; Passini, N.; et al. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain 2008, 131, 3081–3091. [Google Scholar] [CrossRef]
- Heslegrave, A.; Heywood, W.; Paterson, R.; Magdalinou, N.; Svensson, J.; Johansson, P.; Ohrfelt, A.; Blennow, K.; Hardy, J.; Schott, J.; et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 3. [Google Scholar] [CrossRef]
- Piccio, L.; Deming, Y.; Del-Aguila, J.L.; Ghezzi, L.; Holtzman, D.M.; Fagan, A.M.; Fenoglio, C.; Galimberti, D.; Borroni, B.; Cruchaga, C. Cerebrospinal; Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016, 131, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Schlepckow, K.; Kleinberger, G.; Fukumori, A.; Feederle, R.; Lichtenthaler, S.F.; Steiner, H.; Haass, C. An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol. Med. 2017, 9, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Del-Aguila, J.L.; Benitez, B.A.; Li, Z.; Dube, U.; Mihindukulasuriya, K.A.; Budde, J.P.; Farias, F.H.G.; Fernandez, M.V.; Ibanez, L.; Jiang, S.; et al. TREM2 brain transcript-specific studies in AD and TREM2 mutation carriers. Mol. Neurodegener. 2019, 14, 18. [Google Scholar] [CrossRef]
- Suarez-Calvet, M.; Capell, A.; Araque Caballero, M.A.; Morenas-Rodriguez, E.; Fellerer, K.; Franzmeier, N.; Kleinberger, G.; Eren, E.; Deming, Y.; Piccio, L.; et al. CSF progranulin increases in the course of Alzheimer’s disease and is associated with sTREM2, neurodegeneration and cognitive decline. EMBO Mol. Med. 2018, 10, e9712. [Google Scholar] [CrossRef] [PubMed]
- Toden, S.; Zhuang, J.; Acosta, A.D.; Karns, A.P.; Salathia, N.S.; Brewer, J.B.; Wilcock, D.M.; Aballi, J.; Nerenberg, M.; Quake, S.R.; et al. Noninvasive characterization of Alzheimer’s disease by circulating, cell-free messenger RNA next-generation sequencing. Sci. Adv. 2020, 6, eabb1654. [Google Scholar] [CrossRef]
- Koh, W.; Pan, W.; Gawad, C.; Fan, H.C.; Kerchner, G.A.; Wyss-Coray, T.; Blumenfeld, Y.J.; El-Sayed, Y.Y.; Quake, S.R. Noninvasive in vivo monitoring of tissue-specific global gene expression in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 7361–7366. [Google Scholar] [CrossRef]
- Sheinerman, K.S.; Toledo, J.B.; Tsivinsky, V.G.; Irwin, D.; Grossman, M.; Weintraub, D.; Hurtig, H.I.; Chen-Plotkin, A.; Wolk, D.A.; McCluskey, L.F.; et al. Circulating brain-enriched microRNAs as novel biomarkers for detection and differentiation of neurodegenerative diseases. Alzheimers Res. Ther. 2017, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Schor, N.F. What the halted phase III γ-secretase inhibitor trial may (or may not) be telling us. Ann. Neurol. 2011, 69, 237–239. [Google Scholar] [CrossRef]
- Szaruga, M.; Munteanu, B.; Lismont, S.; Veugelen, S.; Horre, K.; Mercken, M.; Saido, T.C.; Ryan, N.S.; De Vos, T.; Savvides, S.N.; et al. Alzheimer’s-Causing Mutations Shift Abeta Length by Destabilizing γ-Secretase-Abetan Interactions. Cell 2017, 170, 443–456.e14. [Google Scholar] [CrossRef]
- Bateman, R.J.; Benzinger, T.L.; Berry, S.; Clifford, D.B.; Duggan, C.; Fagan, A.M.; Fanning, K.; Farlow, M.R.; Hassenstab, J.; McDade, E.M.; et al. The DIAN-TU Next Generation Alzheimer’s prevention trial: Adaptive design and disease progression model. Alzheimers Dement. 2017, 13, 8–19. [Google Scholar] [CrossRef]
- Tariot, P.N.; Lopera, F.; Langbaum, J.B.; Thomas, R.G.; Hendrix, S.; Schneider, L.S.; Rios-Romenets, S.; Giraldo, M.; Acosta, N.; Tobon, C.; et al. The Alzheimer’s Prevention Initiative Autosomal-Dominant Alzheimer’s Disease Trial: A study of crenezumab versus placebo in preclinical PSEN1 E280A mutation carriers to evaluate efficacy and safety in the treatment of autosomal-dominant Alzheimer’s disease, including a placebo-treated noncarrier cohort. Alzheimers Dement. 2018, 4, 150–160. [Google Scholar]
- Reiman, E.M.; Langbaum, J.B.; Fleisher, A.S.; Caselli, R.J.; Chen, K.; Ayutyanont, N.; Quiroz, Y.T.; Kosik, K.S.; Lopera, F.; Tariot, P.N. Alzheimer’s Prevention Initiative: A plan to accelerate the evaluation of presymptomatic treatments. J. Alzheimers Dis. 2011, 26 (Suppl. S3), 321–329. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Voss, T.; Mukai, Y.; Aisen, P.S.; Cummings, J.L.; Tariot, P.N.; Vellas, B.; van Dyck, C.H.; Boada, M.; et al. Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1408–1420. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, S.D.; Che, S.; Counts, S.E.; Mufson, E.J. Shift in the ratio of three-repeat tau and four-repeat tau mRNAs in individual cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. J. Neurochem. 2006, 96, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.; Zhu, J.; Conrad, C.; Schoenfeld, D.; Fang, Z.; Ingelsson, M.; Stamm, S.; Church, G.; Hyman, B.T. Single molecule profiling of tau gene expression in Alzheimer’s disease. J. Neurochem. 2007, 103, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Rosler, T.W.; Carlsson, T.; de Andrade, A.; Fiala, O.; Hollerhage, M.; Oertel, W.H.; Goedert, M.; Aigner, A.; Hoglinger, G.U. Tau silencing by siRNA in the P301S mouse model of tauopathy. Curr. Gene Ther. 2014, 14, 343–351. [Google Scholar] [CrossRef] [PubMed]
- DeVos, S.L.; Miller, R.L.; Schoch, K.M.; Holmes, B.B.; Kebodeaux, C.S.; Wegener, A.J.; Chen, G.; Shen, T.; Tran, H.; Nichols, B.; et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med. 2017, 9, eaag0481. [Google Scholar] [CrossRef] [PubMed]
- Duffy, A.G.; Makarova-Rusher, O.V.; Ulahannan, S.V.; Rahma, O.E.; Fioravanti, S.; Walker, M.; Abdullah, S.; Raffeld, M.; Anderson, V.; Abi-Jaoudeh, N.; et al. Modulation of tumor eIF4E by antisense inhibition: A phase I/II translational clinical trial of ISIS 183750-an antisense oligonucleotide against eIF4E-in combination with irinotecan in solid tumors and irinotecan-refractory colorectal cancer. Int. J. Cancer 2016, 139, 1648–1657. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Painter, M.M.; Bu, G.; Kanekiyo, T. Apolipoprotein E as a Therapeutic Target in Alzheimer’s Disease: A Review of Basic Research and Clinical Evidence. CNS Drugs 2016, 30, 773–789. [Google Scholar] [CrossRef]
- Cramer, P.E.; Cirrito, J.R.; Wesson, D.W.; Lee, C.Y.; Karlo, J.C.; Zinn, A.E.; Casali, B.T.; Restivo, J.L.; Goebel, W.D.; James, M.J.; et al. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 2012, 335, 1503–1506. [Google Scholar] [CrossRef]
- Luz, I.; Liraz, O.; Michaelson, D.M. An Anti-apoE4 Specific Monoclonal Antibody Counteracts the Pathological Effects of apoE4 In Vivo. Curr. Alzheimer Res. 2016, 13, 918–929. [Google Scholar] [CrossRef]
- Liao, F.; Li, A.; Xiong, M.; Bien-Ly, N.; Jiang, H.; Zhang, Y.; Finn, M.B.; Hoyle, R.; Keyser, J.; Lefton, K.B.; et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Investig. 2018, 128, 2144–2155. [Google Scholar] [CrossRef] [PubMed]
- Mungenast, A.E.; Siegert, S.; Tsai, L.H. Modeling Alzheimer’s disease with human induced pluripotent stem (iPS) cells. Mol. Cell. Neurosci. 2016, 73, 13–31. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Pan, Y.; Short, J.L.; Choy, K.H.; Zeng, A.X.; Marriott, P.J.; Owada, Y.; Scanlon, M.J.; Porter, C.J.; Nicolazzo, J.A. Fatty Acid-Binding Protein 5 at the Blood-Brain Barrier Regulates Endogenous Brain Docosahexaenoic Acid Levels and Cognitive Function. J. Neurosci. 2016, 36, 11755–11767. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, P.; Gogia, N.; Singh, A. Exploring the efficacy of natural products in alleviating Alzheimer’s disease. Neural Regen. Res. 2019, 14, 1321–1329. [Google Scholar] [PubMed]
- Sarkar, A.; Irwin, M.; Singh, A.; Riccetti, M.; Singh, A. Alzheimer’s disease: The silver tsunami of the 21(st) century. Neural Regen. Res. 2016, 11, 693–697. [Google Scholar] [PubMed]


{kind=link}
| Approaches to Risk Prediction | |||||||
|---|---|---|---|---|---|---|---|
| Ref. | Approach | Findings | |||||
| [127] | PRS | EOAD, sLOAD, and fLOAD have different PRS profiles | |||||
| [128] | PRS + biomarker | Prediction of conversion or AAO | |||||
| [129] | PRS + brain atrophy + MMSE score | Better progression prediction | |||||
| [130] | PRS + brain atrophy + MMSE score + CSF data | Individuals with high PRS and with amyloid and tau pathology showed a faster rate of memory decline, even among APOE ε4 non-carriers | |||||
| [131] | PRS | PRS differentiate AD, FTD, PD, and ALS | |||||
| Biomarkers for Early Diagnosis | |||||||
| Ref. | Target | Localization | Indicative Of | Aβ-Independent | Tau-Independent | ||
| [132] | sTREM2 | CSF, plasma | Onset and progression of tau pathology | Y | N | ||
| [133] | Nfl | plasma, CSF | Cytoskeleton protein released with neuron death | Y | Y | ||
| [134] | sPDGRβ | blood | Blood-brain barrier breakdown | Y | Y | ||
| Possible Treatments and Clinical Trials | |||||||
| Clinical Trial Ref. | Target | Mechanism | Participants | Goal | Drug | Status | |
| NCT01677572 | Amyloid β | Monoclonal antibodies | Mild AD and MCI | Clearance of Aβ plaques | Aducanumab | Approved to treat Alzheimer’s disease | |
| NCT02760602 | Prodromal AD | Solanezumab | T—no evidence that prodromal AD benefits from drug | ||||
| NCT02008357 | Older Individuals at risk (APOE4+) | Solanezumab | P3—not recruiting | ||||
| NCT01760005 | DIAN-TU | Solanezumab | P3; H—no change in cognitive performance | ||||
| NCT01760005 | DIAN-TU | Gantereumab | P3; H—no change in cognitive performance | ||||
| NCT01998841 | Colombian family | Crenezumab | P3—not recruiting | ||||
| NCT01661673 | Y-secretase | y-modulator | Mild Cognitive Impairment | Decrease production of toxic Aβ | EVP-0962 | P2—completed | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibanez, L.; Cruchaga, C.; Fernández, M.V. Advances in Genetic and Molecular Understanding of Alzheimer’s Disease. Genes 2021, 12, 1247. https://doi.org/10.3390/genes12081247
Ibanez L, Cruchaga C, Fernández MV. Advances in Genetic and Molecular Understanding of Alzheimer’s Disease. Genes. 2021; 12(8):1247. https://doi.org/10.3390/genes12081247
Chicago/Turabian StyleIbanez, Laura, Carlos Cruchaga, and Maria Victoria Fernández. 2021. "Advances in Genetic and Molecular Understanding of Alzheimer’s Disease" Genes 12, no. 8: 1247. https://doi.org/10.3390/genes12081247
APA StyleIbanez, L., Cruchaga, C., & Fernández, M. V. (2021). Advances in Genetic and Molecular Understanding of Alzheimer’s Disease. Genes, 12(8), 1247. https://doi.org/10.3390/genes12081247