1. Introduction
CRISPR-Cas technology has revolutionized the gene editing space, enabling access to the field for many new users [
1]. Due to its versatility and ease of use, it is being deployed for usage in vivo as a method to treat genetic disorders requiring novel delivery methods and a means to monitor gene editing in vivo [
2]. For example, somatic in vivo CRISPR–Cas9 gene editing was recently performed in humans as a treatment for Leiber’s congenital amaurosis 10 (LCA10) [
3].
Previous methods to monitor in vivo CRISPR editing have commonly relied on nucleotide sequencing of edited target biopsies or invasive monitoring of the tissues for fluorescent markers [
4]. These methods require the destruction of the tissue of interest and prevent long term serial monitoring of CRISPR editing. Additionally, without non-invasive monitoring, unpredicted off-target tissue activity may not be detected, unless each tissue is tested directly. Therefore, a method with the ability to monitor CRISPR activity non-invasively is an important tool for developing gene editing delivery systems.
The cre-
loxP system is also a powerful tool for animal and cell models, providing robust and specific editing of DNA [
5]. Cre recombinase is derived from bacteriophage P1. Cre recombines DNA contained within
loxP sequences, creating inversions, deletions, and insertions depending on the orientation of the
loxP sites [
6]. Thousands of animal models have been generated using cre-
loxP technologies, 320 of which are specifically mouse reporter models [
7]. Many cre-
loxP animal models have also been developed in which fluorescent or luminescent reporter genes are inactivated by upstream “floxed” sequences, which are two
loxP sites surrounding a polyadenylation (polyA) “stop” sequence or another gene [
8,
9]. When the cells of these animals are exposed to cre, the floxed sequence is excised, this activates reporter gene expression.
The Barry lab previously made use of these floxed reporter mice to “fingerprint” gene delivery in vivo by adeno-associated virus (AAV) vectors [
10]. In this approach, AAV-cre was injected into mice that are transgenic with different cre-activated reporter genes. In LSL-luciferase mice, luciferase’s expression is blocked by a floxed polyA cassette upstream of luciferase (
Figure 1). In the absence cre, no luciferase is expressed. When cre is delivered, the recombination process results in the net deletion of the floxed stop cassette consisting of neomycin and a polyA transcriptional stop, resulting in an activated luciferase reporter. In mT/mG mice, a floxed membrane-targeted red fluorescent protein mTomato (mT) is followed by membrane-targeted GFP (mG). In the absence of cre, mT is expressed in all cells of the mouse and is membrane targeted. When cre is delivered, mT is deleted and mG is expressed. At higher magnifications, these membrane-targeted reporter proteins provide substantial cell discrimination [
8].
More recently, a similar “footprinting” system was used to track gene delivery with cre recombinase and notably Cas9 by monitoring fluorescent protein activation [
11]. This approach lacked the ability to track editing in living animals by luciferase imaging but had the added value of tracking Cas9 in vivo post-mortem by tissue sectioning. In this case, Cas9 was targeted by gRNA to a polyA “stop” cassette near
loxP sites in mice bearing floxed inactivated fluorescent protein genes. This design is limited to the specific stop cassette in this mouse model and is not widely applicable to other mouse reporter models. Cas9 cleavage followed by host cell DNA repair was able to activate reporter gene expression to monitor in vivo editing [
11].
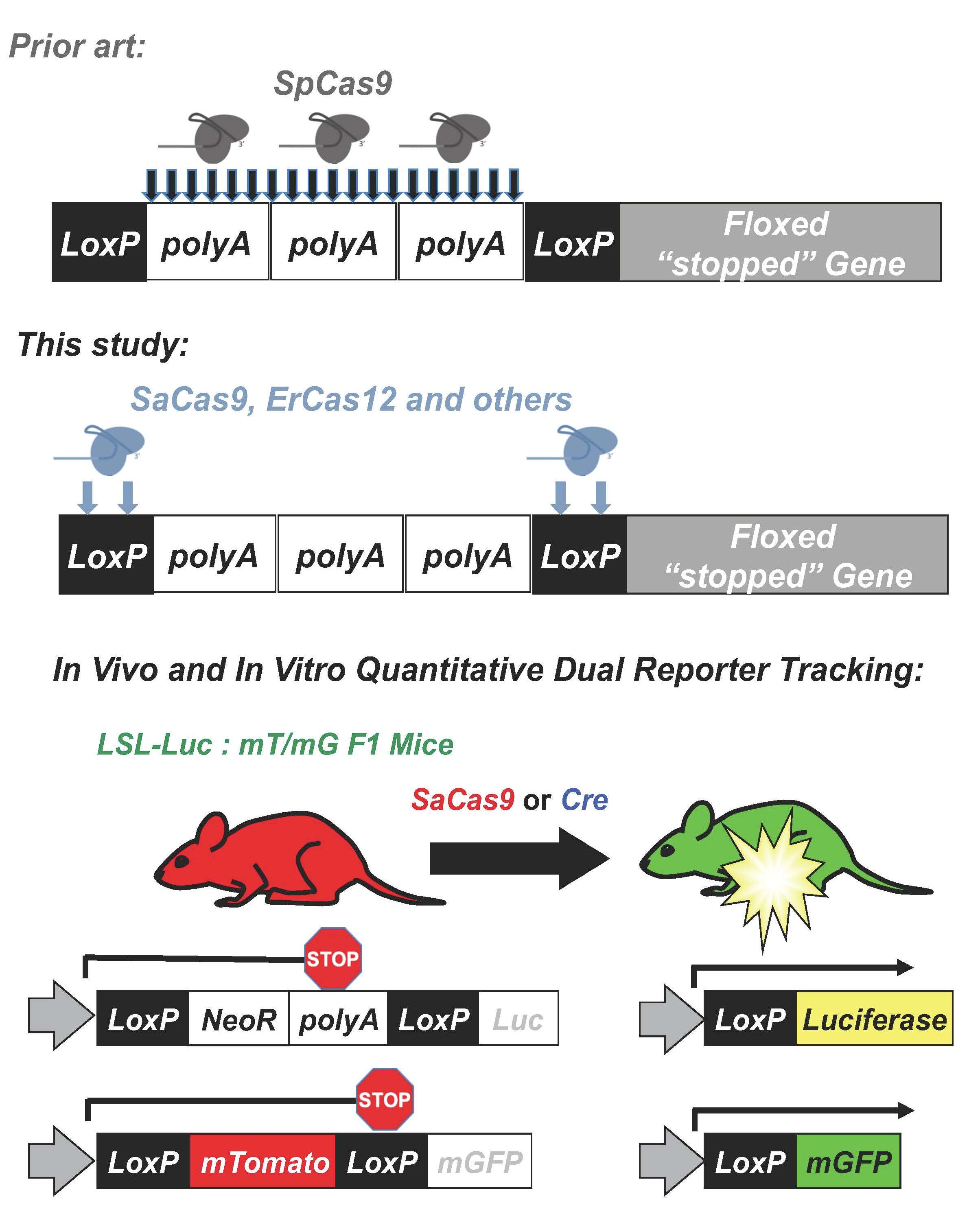
While this footprinting approach was novel, its targeting of the polyA sequence will work only in a small set of 10 fluorescent and neural mouse models that have the same stop cassette [
12]. Mice that have stop cassettes that express a gene rather than repeated polyAs as the stop cassette, like the LSL-luciferase and mT/mG mice (see Graphical Abstract), cannot use a polyA based gRNA. This is because the gRNAs will not create a large deletion and can even have off-target editing of the similar target sites. Additionally, if making a hybrid mouse with 2 different
loxP cassettes, the only site guaranteed to be an editable target on both chromosomes would be
loxP.
Given this, CRISPR-Cas9 was targeted to loxP sequences and tested in the combined luciferase and mT/mG mouse model for combined live imaging and post-mortem evaluation of genome editing. By doing this, it was shown that SaCas9 activation of these reporter genes has near equivalent activity to that of cre recombinase, in vitro and in vivo. Through sequencing, a primary pathway was not conclusively determined due to ambiguity of the repair outcomes, although non-homologous end-joining events were amongst the top reads. Finally, the targeting of loxP with SaCas9 and other CRISPR nucleases has opened the door to using the plethora of established loxP models to test new delivery methods and monitoring for CRISPR gene editing activity in vivo.
2. Materials and Methods
2.1. Plasmids and Cloning
The reporter plasmid p133 pSV-STOP-luciferase was a gift from Jeffrey Green (Addgene plasmid #8390;
http://n2t.net/addgene:8390; RRID: Addgene_8390, accessed on 15 April 2020) and consisted of an SV40 promoter upstream of a floxed set of SV40 polyA signals with firefly luciferase downstream of the floxed region (Addgene, Watertown, MA, USA). Plasmid px601 was purchased from Addgene and consisted of a CMV-expressed SaCas9 and gRNA expression cassette flanked by AAV ITRs (pX601-AAV-CMV::NLS-SaCas9-NLS-3xHA-bGHpA;U6::BsaI-sgRNA was a gift from Feng Zhang (Addgene plasmid #61591;
http://n2t.net/addgene:61591, accessed on 15 April 2020; RRID: Addgene_61591)). Annealed oligos were inserted into the gRNA cassette after plasmid px601 was digested with BsmBI. This method was previously described by Ran et al. [
13]. The plasmids that were successfully cloned with the
loxP gRNAs 1 and 2 and were named plasmid px601
loxP1 and plasmid px601
loxP2, respectively. pSC-CMV-cre was previously developed by the Barry lab and consisted of a CMV-driven cre flanked by AAV ITRs [
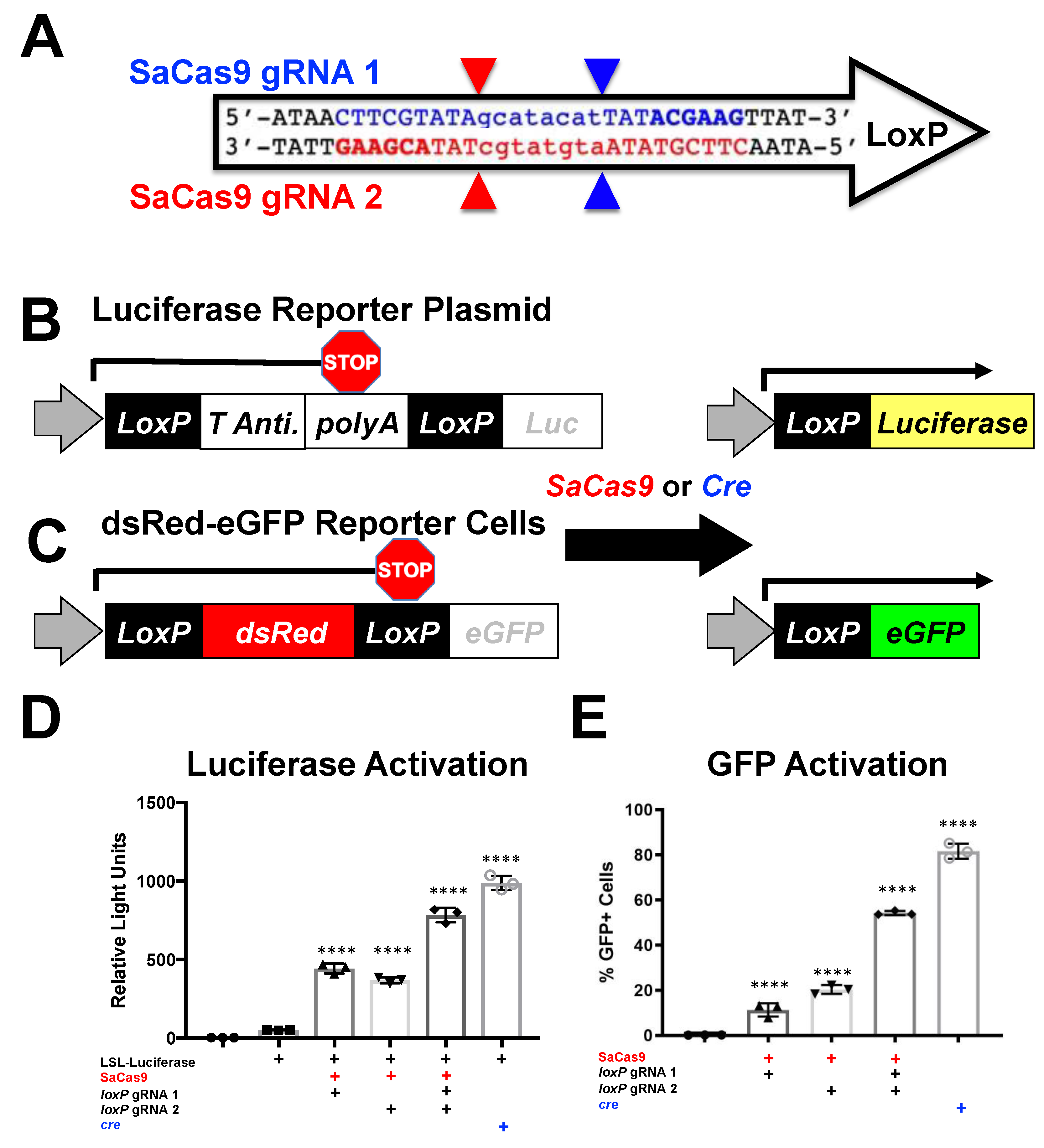
10]. ErCas12a plasmid was cloned as previously described in Wiersen et al. [
14].
2.2. Cell Culture
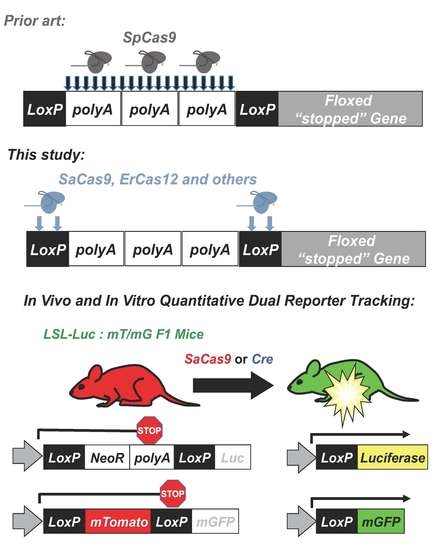
HEK293 cells purchased from ATCC (ATCC
® CRL-1573™) were grown with Dulbecco’s modified Eagle’s medium completed with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and penicillin–streptomycin at 100U/mL (Thermo Fisher Scientific). Plasmid transfections were done with Xfect Transfection Reagent from Takara Bio into 6-well plates of HEK293 cells when the cells were at 70% confluency following manufacturer’s instructions. Transfections consisted of 2.5 µg of editing plasmids (px601 gRNA1, px601 gRNA2, and pSC-CMV-cre) and 2.5 µg of reporter plasmids. Transfections that used two gRNAs used 1.25 µg of px601
loxP1 and px601
loxP2 each. Cells were harvested 48 h post-transfection for luciferase assays. HEK293 cells were transduced with a lentivirus constructed from pLV-CMV-
loxP-DsRed-
loxP-eGFP and selected for using the puromycin selection marker. pLV-CMV-LoxP-DsRed-LoxP-eGFP was a gift from Jacco van Rheenen (Addgene plasmid # 65726;
http://n2t.net/addgene:65726, accessed on 15 April 2020; RRID:Addgene_65726) [
15]. Once these were established, the cells acted as the reporter system in
Figure 1C and had the activity shown in
Figure 1E (the IRES puromycin selection cassette downstream of the fluorescent cassettes is not shown). The cells were transfected with 5 μg of px601
loxP1, 5 μg of px601
loxP2, and 2.5 μg of each or with 5 μg of cre reporter plasmid. For the ErCas12a analysis, cells were plated into a 6-well plate and transfected at 60–80% confluency with Xfect and 2.5 μg of the reporter plasmid was transfected to be targeted by ErCas12a. The ErCas12a plasmid co-expressed either gRNA 1 or 2 and was co-transfected at 2.5 μg. For the cells transfected with both gRNAs, 1.25 μg of each was transfected into that well. Cells were harvested for a luciferase assay. By one-way ANOVA, all the groups were significant except between the untransfected control group and the P133-transfected group and between the individual gRNA-treated groups; 95% confidence intervals are shown (
n = 3).
2.3. Luciferase Assay
Cells were harvested from each condition via trypsin. The cells were spun down at 500× g for 10 min and the supernatant was removed. The cells were resuspended in 400 µL of PBS and pipetted vigorously to have a homogenous mixture of cells. Then, 100 µL of cells was pipetted into a clear bottom, black, 96- well plate. This was done in triplicate for each group of cells and 100 µL of room temperature Bright-Glo was then applied to each well. Wells were then analyzed via the BioTek Synergy H1 Microplate Read and the BioTek Gen5 Microplate Software.
2.4. Flow Cytometry
Cells were harvested as with the luciferase assay. The cells were passed through a 100 μm filter to avoid clumping of cells. The cells were then assayed by the Microscopy and Cell Analysis Core at Mayo Clinic Rochester (Rochester, Minnesota, USA) using a BD FACSCanto. The data were then processed via FloJo.
2.5. Mice
LSL-luciferase mice (a.k.a. FVB.129S6(B6)-GT(ROSA)26Sortm1(Luc)Kael/J) have a luciferase expression cassette behind a floxed polyA signal domain with the ROSA26 promoter. The mT/mG mice (a.k.a. GT(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J mice) have a floxed mTomato behind the pCA promoter with membrane-bound EGFP behind mTomato. All mice were purchased from Jackson Laboratories and all animals were treated according to the provisions of the Animal Welfare Act, PHS Animal Welfare Policy, and NIH Guide.
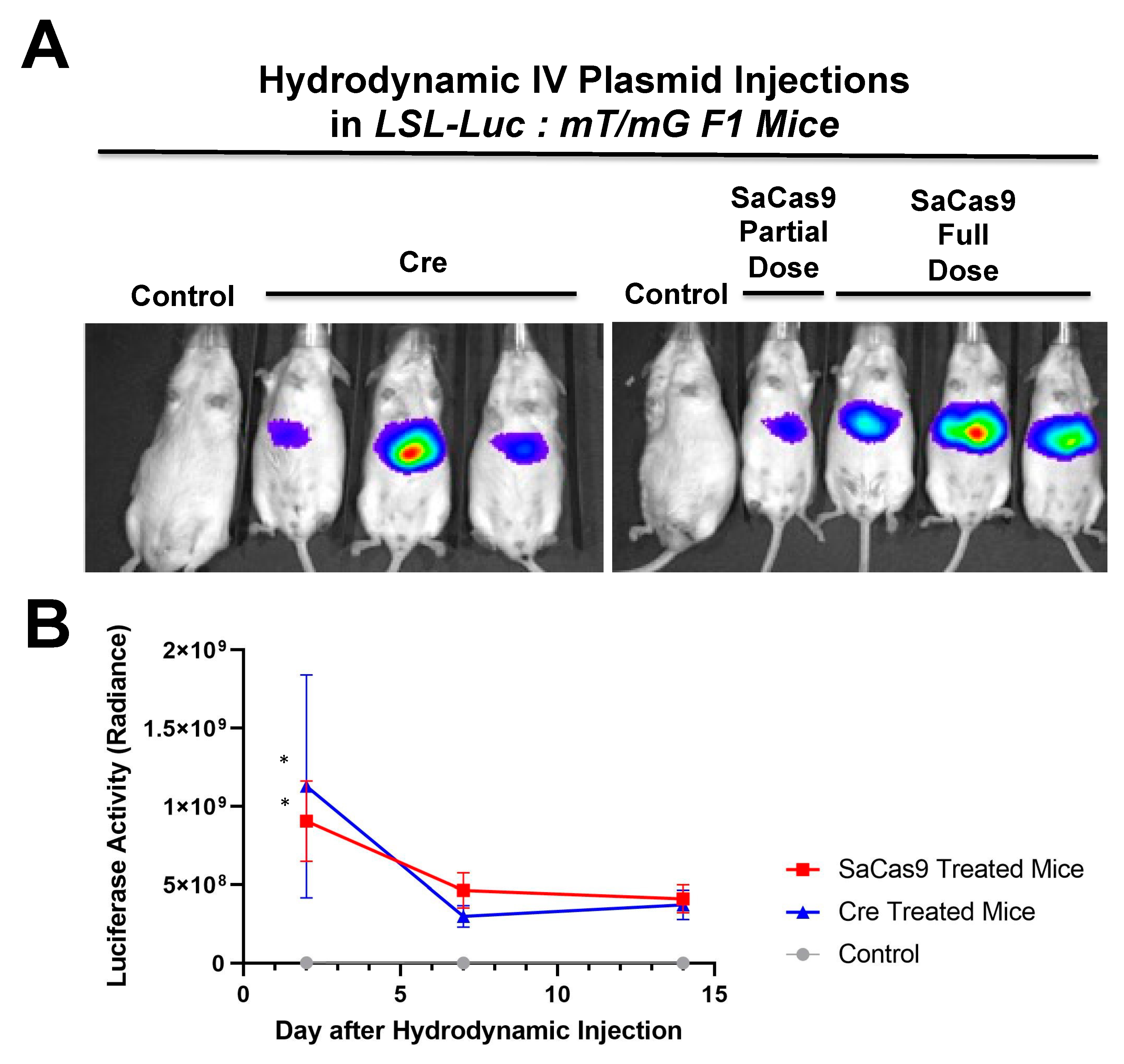
2.6. Hydrodynamic Injections
Mice were injected by hydrodynamic tail vein injections [
16]. Injections consisted of 25 µg of gene editing plasmids, either px601
loxP1 and px601
loxP2 mixed in a 50:50 ratio or pSC-CMV-cre. The plasmids were suspended in 2.5 mL (~10% body weight) of PBS and the injection was completed within 8–10 s [
16]. Control mice were injected with PBS only. Mice were monitored post-injection for tolerance of hydrodynamic injection. For the dose escalation hydrodynamic injections, 7.5 μg, 25 μg, and 83.3 μg of plasmid were used for the low, medium, and high groups, respectively. Pilot experiments were conducted with intramuscular soleus injections of 100 μg of plasmid in 50 µL of PBS (
Supplementary Figure S3).
2.7. In Vivo Bioluminescent Imaging
Mice were anesthetized with 2% isofluorane and maintained at 2% isofluorane. Mice were intraperitoneally injected with 150 µL of 20 mg/mL D-Luciferin (RR Labs, Inc., San Diego, CA, USA). Xenogen IVIS 200 was used to image the mice ten minutes after injection of the D-Luciferin. The dose escalation imaging experiments were done using the IVIS Lumina S5 Imaging System due to the Xenogen being retired. Living Image software was used to quantify the luminescence.
2.8. Liver Sectioning and Confocal Microscopy of Mouse Liver
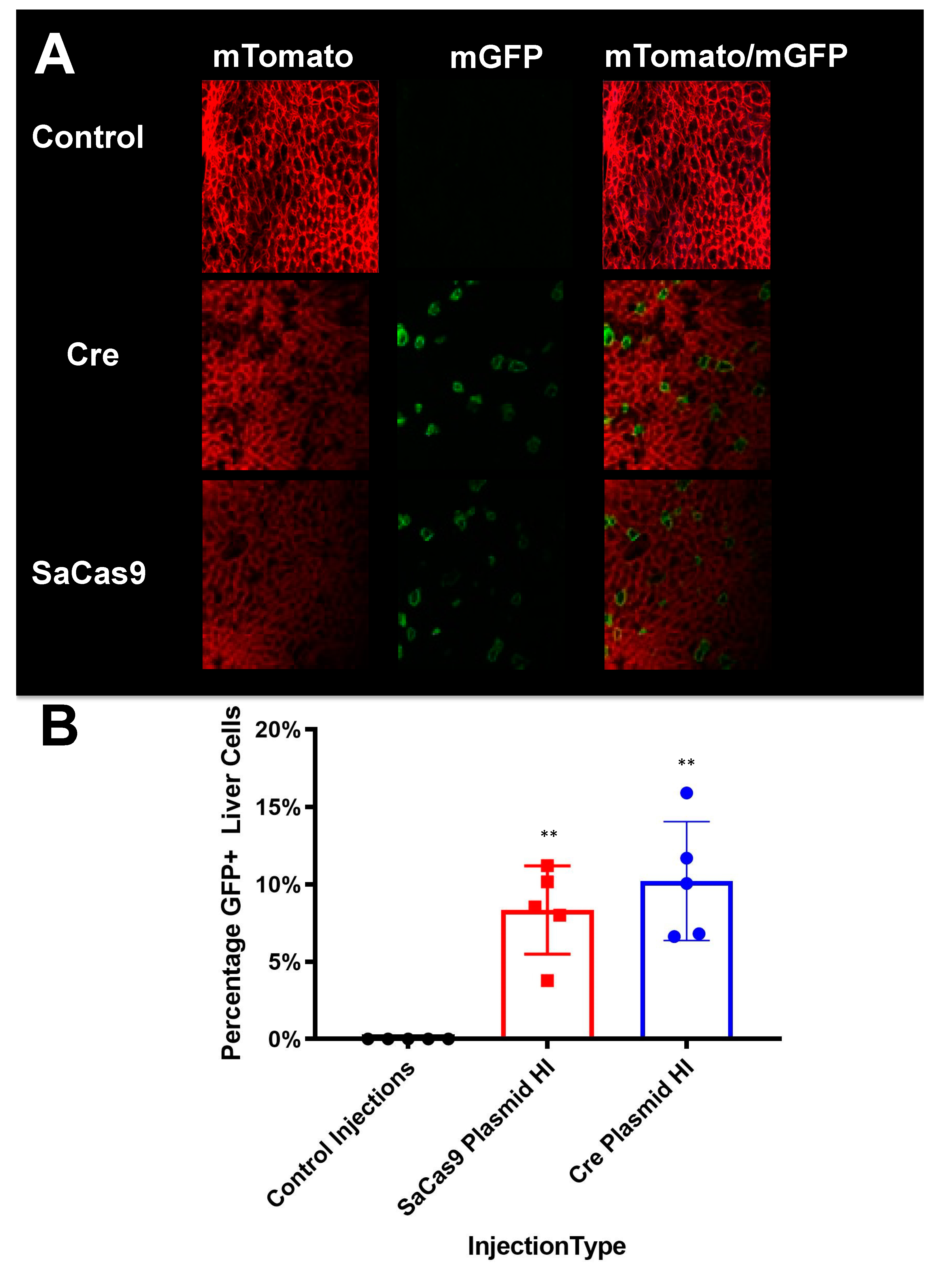
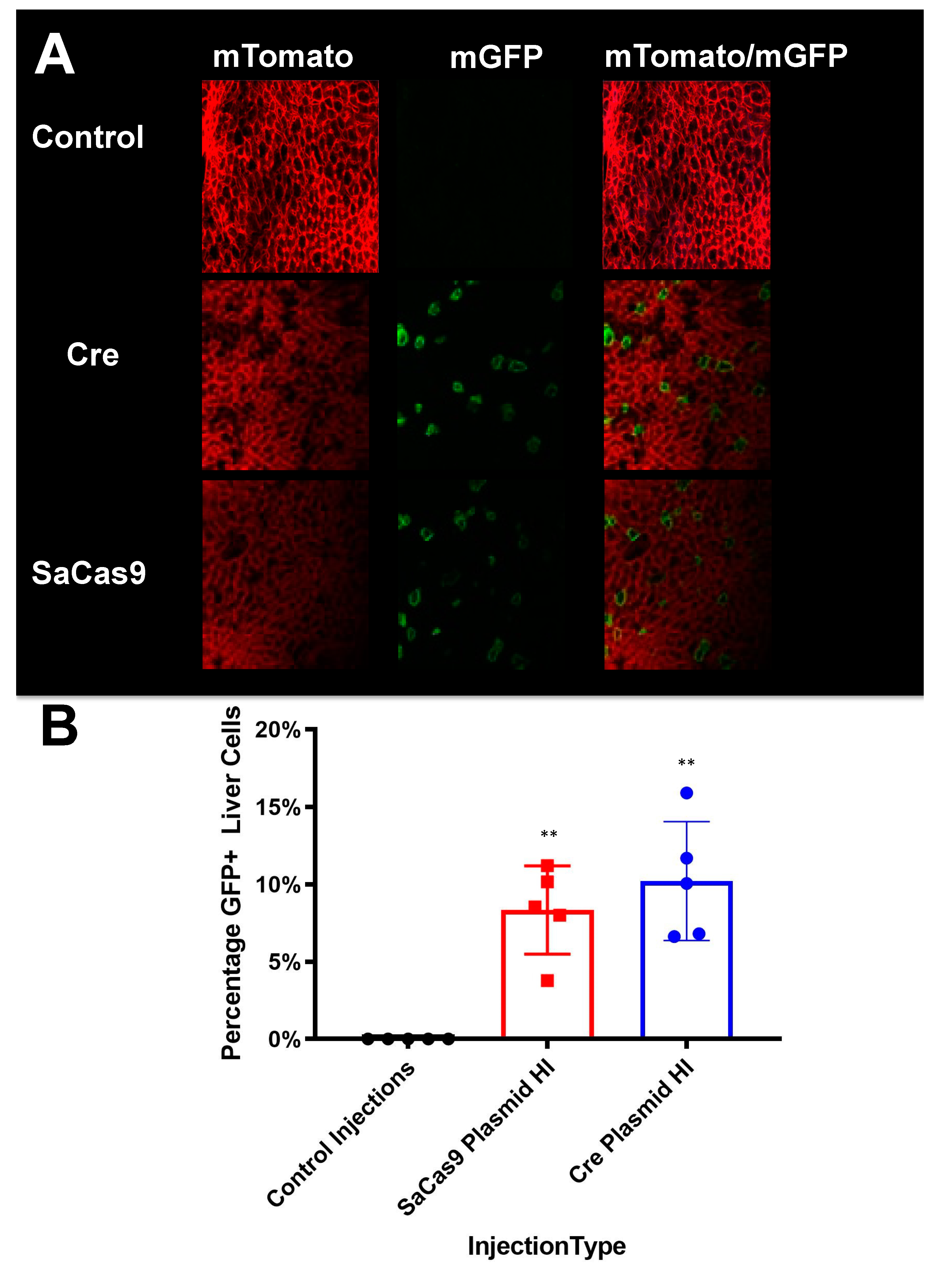
Livers were harvested and fixed overnight in 4% paraformaldehyde (PFA)-phosphate buffered saline (PBS) at 4 °C. Livers were transferred to 15% sucrose–PBS overnight and then moved to 30% sucrose–PBS at 4 °C until the tissue sunk in the solution to cryoprotect the tissue. Tissues were trimmed and flash frozen in optimal cutting temperature (OCT) medium (Sakura Finetek USA, Torrance, CA, USA). A Leica CM1860 UV cryostat (Leica Biosystems) was used to create cryosections (18 μm thickness) which were mounted on slides (SuperFrost Plus; Thermo Fisher Scientific, Waltham, MA). VECTASHIELD with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA) was applied with a CytoSeal-60 coverslip sealant (Thermo Fisher Scientific). The confocal microscopy was performed at the Microscopy and Cell Analysis Core facility at Mayo Clinic Rochester (Rochester, MN, USA) using a Zeiss LSM790 laser confocal microscope (Carl Zeiss Jena, Jena, Germany). Representative images were selected from the confocal microscopy and used to count GFP+ cells and total cells.
2.9. Sequencing of loxP Junctions
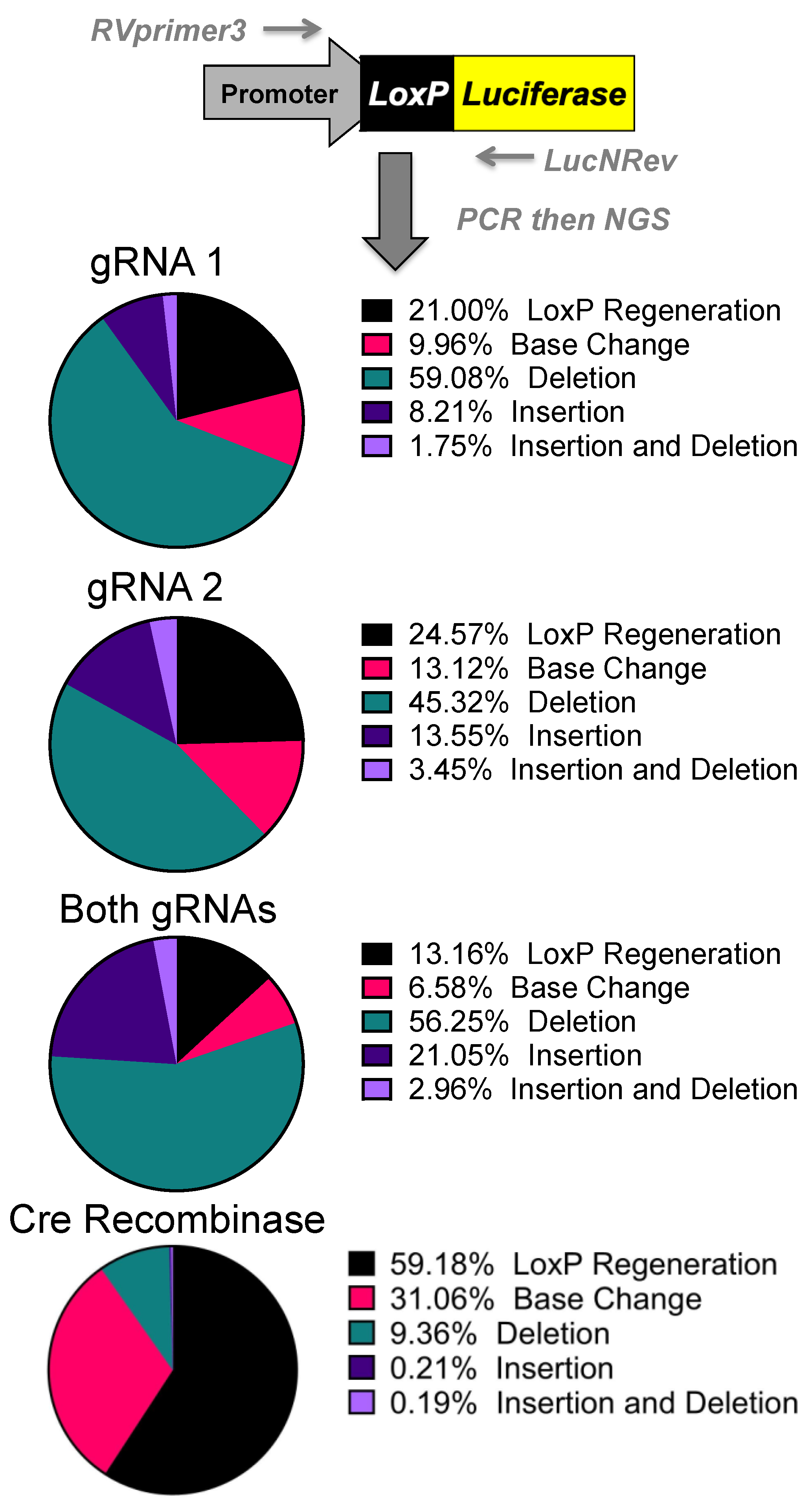
DNA was isolated from the transfected human 293 cells via the DNeasy Blood and Tissue Kit’s protocol. The primers RVprimer3 (5′-CTAGCAAAATAGGCTGTCCC-3′) and LucNRev (5’-CCTTATGCAGTTGCTCTCC-3′) were used to PCR amplify from the SV40 promoter across the polyA region or the deleted region. Shortening the extension time to 30 s restricts the amplified product to only edited plasmids. This band was then gel-excised and stratacloned to isolate individual sequences. Individual clones were grown up, miniprepped, and sent for Sanger sequencing at Genewiz. Additionally, gel-isolated bands were also sent for next generation sequencing (NGS) via the Genewiz’s Amplicon EZ sequencing using the extended primers RVprimer3 NGS (5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCTCTAGCAAAATAGGCTGTCCC-3′) and LucNRev NGS (5′-GACTGGAGTTCAGACGTGTGCTCTTCCGATCT CCTTATGCAGTTGCTCTCC-3′). Read outcomes were compiled and ranked from most common to least. This method creates ~50,000 reads of the 500bp fragments. This is a biased analysis that will only monitor large deletions of the stop cassette and does not quantify unedited plasmids. These reads were then aligned to the predicted loxP regeneration that one would expect from cre recombinase or a one gRNA NHEJ event. This system is set up to monitor mutations at the site of interest and Genewiz sends the reads analyzed with WT, base change, deletion, insertion, and insertion/deletion. WT is the loxP regeneration site and this shows the site has recombined into the predicted site for cre recombination with the loxP sites being recombined. Base change is a loxP regeneration site with a base change within 10bp of the loxP site. “Deletion” and “insertion” are as they sound, deletion or insertion, compared to the loxP regeneration site and “insertion and deletion” denote reads that show signs of both. The top five outcomes were looked at for potential connection to a DNA repair pathway, but due to the potential for loxP regeneration to be the result of any repair pathway, this was not pursued.
2.10. Statistical Analysis
T-test and one-way ANOVA with multiple comparisons were done using GraphPad Prism™. A 95% confidence interval was displayed on the graphs and those considered significant by one-way ANOVA with Tukey’s multiple comparison were marked as significant. Significance between groups is described in the figure legends in more detail between the groups and beside the control groups.
4. Discussion
This study was performed to enable a facile system for monitoring genome editing in living animals as well as to identify edited tissue at a cellular resolution. This work shows that established cre-loxP reporter systems can be used to monitor CRISPR activity. This paper showed that loxP cleavage by SaCas9 can be comparable to cre recombinase activity in deleting stop signals in vitro or in vivo. By using the previously established mT/mG:LSL Luc, it was demonstrated that CRISPR activity could be detected non-invasively in living mice along with a quantitative approach upon dissection of edited tissues.
This system also establishes the first
loxP specific reporter for fluorescence monitoring. Previous work has used gRNAs to target adjacent to the
loxP site or within the polyA region (
Supplementary Figure S2A). While these have shown the ability to monitor CRISPR activity via tissue sectioning and DNA analysis, these systems are specific to Ai9 mice and a few related strains and would not be broadly applicable to other reporter systems such as the LSL-mice or the mT/mG target loci [
25]. Other work has shown in vitro targeting with partial overlap with
loxP and complete coverage with the gRNA in the
loxP mutants
lox71 and
lox66 (
Supplementary Figure S2B) [
26]. These previous gRNAs would not be applicable to most mouse models that likely will not have convenient adjacent sequences that allow for targeting and most mouse reporter models do not use
loxP mutants. The system shown herein can be used with any wildtype LoxP site and is therefore capable of being used with any mouse model that uses
loxP to activate or deactivate genes by cre excision. This system could also be used in cases where
loxP is positioned for gene inversion to delete a region or disrupt this process.
Cre recombinase has evolved to not only cut
loxP sequences, but to also repair the resulting genomic lesion. In contrast, Cas editors simply cleave DNA. Any Cas cleavage event must rely on the host cell to repair the cut DNA. Therefore, one might assume that Cas-mediated deletion of a floxed locus would likely be less efficient than the evolved cut and repair cre recombinase system. However, when using SaCas9 and the dual guide system, SaCas9 was like cre in efficiency for reporter activation in vitro and in vivo (
Figure 1,
Figure 4 and
Figure 5). This activity level can be partially explained by SaCas9, an efficient DNA editor that can show higher activity than SpCas9 and a Cas12a variant when using similar or identical targets [
21].
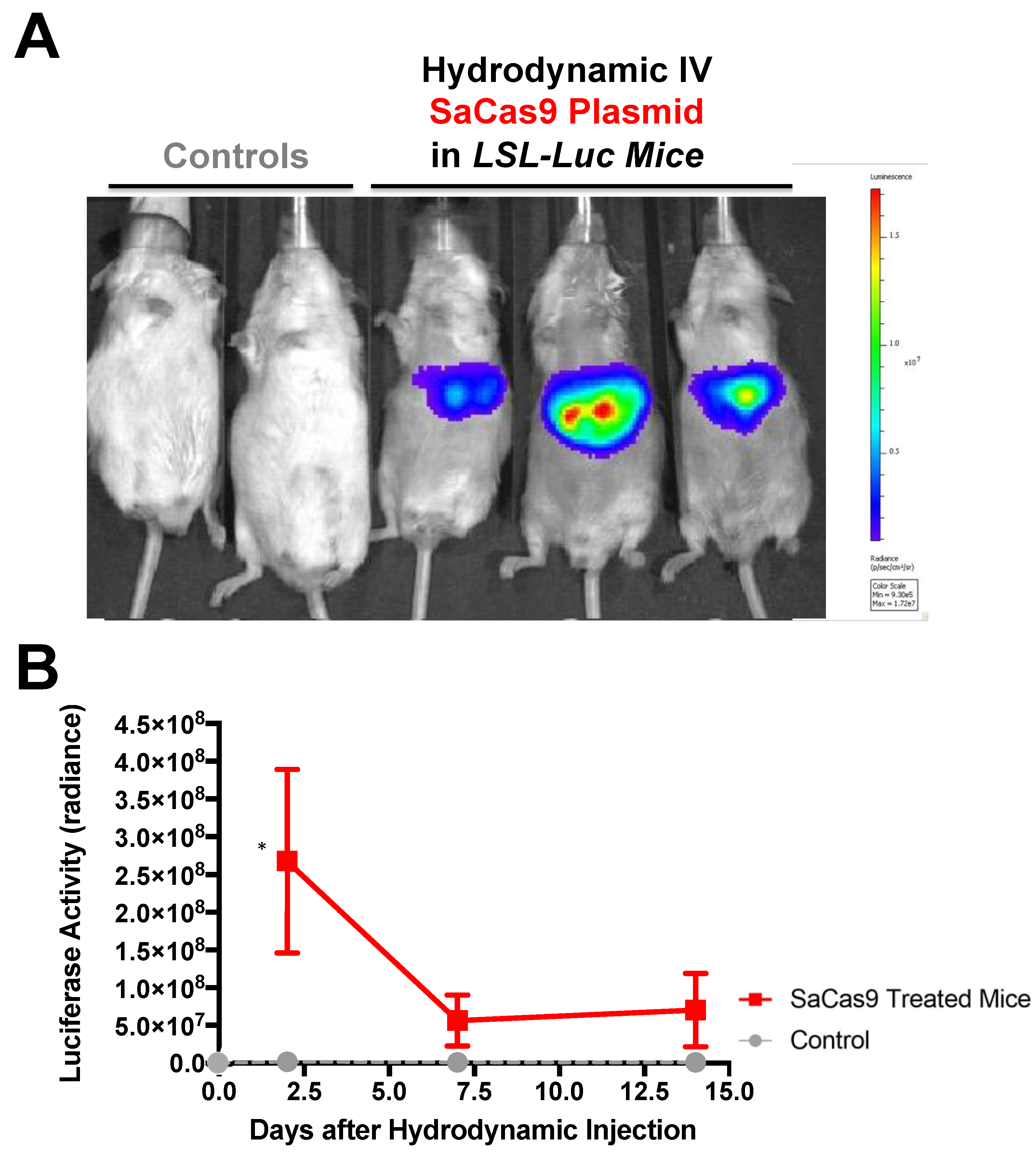
Hydrodynamic delivery functions by rupturing the cell membranes under pressure, pushing the plasmid DNA into the cell. This pressure and resulting damage could be responsible for the decline in luciferase activity from Day 2 to Day 7 as seen in
Figure 3,
Figure 4 and
Figure 6. This coupled with the introduction of neo-antigens in the form of SaCas9 or cre could lead to immunological responses to transduced cells as well. Additionally, there is a discrepancy between the SaCas9 hydrodynamically injected mice in
Figure 4;
Figure 5. Older mice are not as effective for hydrodynamic delivery and older mice were used in
Figure 4. Different maxi-preps were used between the experiments so this may have also contributed.
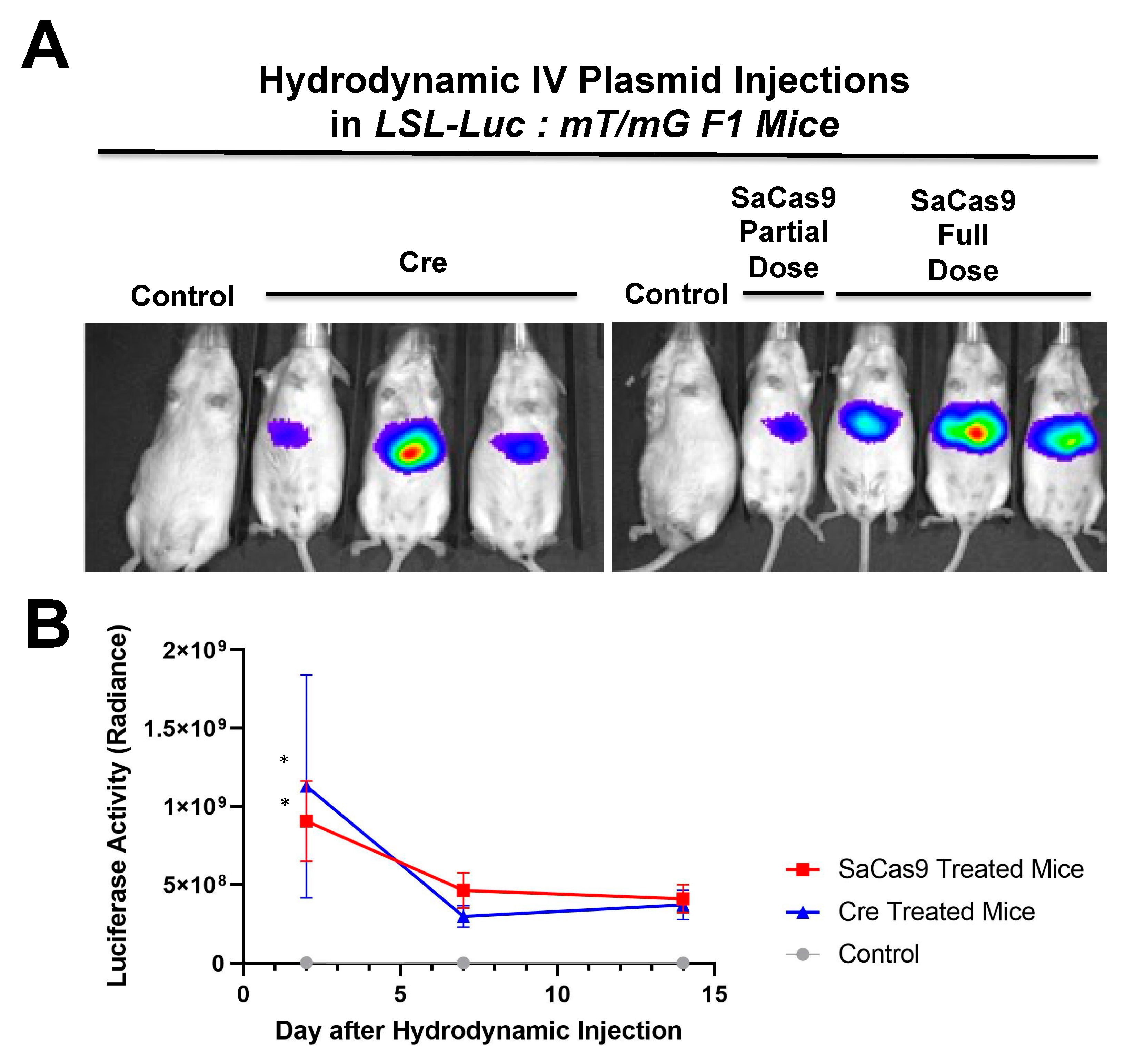
Additionally,
Figure 7 shows that there is a dose response to differing amounts of SaCas9. This is of value because it shows that this system could be used to monitor variant levels of delivery via luciferase activity. Whether SaCas9 is delivered by nanoparticle, virus, ribonucleoprotein, or some novel method, this provides a method to detect variations in delivery efficiency non-invasively and with cellular specificity. It should also be noted that the luciferase levels showed a significant decrease compared to previous experiments. We believe this to be the result of imaging differences between the Xenogen and the Lumina.
Supplementary Figure S3 also shows that this reporter system works in tissues beyond the liver. The Barry lab has also previously published in Hillestad et al. that they were able to detect cre recombinase activity using this reporter mouse in the liver, heart, lungs, muscles, brain, kidney, and spleen [
10].
Next generation sequencing analyses of the gene editing outcomes provide some insight into how the cell repairs these DNA breaks prior to reporter activation. Three repair pathways were non-homology-induced repairs, small local homology (microhomology), and homology-based outcomes, especially considering that deletions occur between two identical
loxP sequences. Analyses of deletions induced by individual gRNAs demonstrated that most of the edits result in the recreation of a
loxP site. Unfortunately, these can be the result of either NHEJ or homology-based repair when using individual gRNAs (
Supplementary Figure S1).
Analysis of the dual gRNA-induced deletions demonstrated a different mix of outcomes (
Supplementary Figure S1). After the repair pathway-ambiguous
loxP recreation, the next two top reads consisted of NHEJ-based events that are caused by different gRNAs targeting each
loxP site. Although the top reads are ambiguous, the top reads that are capable of being definitively linked to a pathway are NHEJ. Depending on which gRNA edits which
loxP site will determine whether an 8bp region would be duplicated or deleted. That being said, these cuts have the potential to cause microhomology-based events, only one of which is distinguishable as such. The strong prevalence of NHEJ-specific events strongly suggests that this is a major repair mechanism, but with the top detected outcome being ambiguous and repeated cutting of regenerated
loxP sites confusing the data further, it cannot be determined at this time what is the primary repair pathway. Further parsing of the mechanism may be done in future work by specifically knocking down proteins related to these pathways.
The sequencing was initially done to explore the possibility of alternative DNA repair pathways being responsible for the increased efficacy of both gRNAs over individual gRNAs. While repair mechanisms cannot be determined conclusively, the increases in efficacy seen with both gRNAs are potentially connected with CRISPR gene editing selecting for mutations that prevent further cutting. With a single gRNA, there is the potential for the gRNA target site to be cut and result in an insertion or deletion (indel) rather than result in a large deletion and preclude the possibility for a subsequent DSB generating a large deletion. An indel near the cut site of the gRNA target site would greatly reduce, if not inhibit, SaCas9 cutting as seen by some of the top reads in NGS [
27]. If a
loxP site is mutated to prevent CRISPR cutting at both
loxP sites, large deletion of the stop cassette would be prevented. With two potential gRNA targets, the potential indel would be further away from the PAM and less likely to inhibit SaCas9 binding [
28]. This may potentially explain the increased efficacy seen with two gRNAs rather than one.
By targeting Cas9 to the loxP site, there is also the opportunity to enable further manipulation of these sites. These models can be used in conjunction with targeted insertion technology to deliver genes of interest at loxP sites. This could be done to modify the sites to express a different gene or to reconstitute stop cassettes with mutant loxP sites that are resistant to CRISPR cutting but available to cre recombinase. There is also the potential of creating a three-outcome cassette: starting cassette, cre recombinase-treated expression cassette, and CRISPR-treated cassette. This would give greater control over animal models that would be able to turn on defective genes via cre recombinase and then deactivate them by CRISPR. There is also the potential of using the deactivated CRISPR enzyme as an inhibitor of cre recombinase, binding loxP and preventing binding and recombination. This could act to prevent cre recombinase activity in specific cell populations.
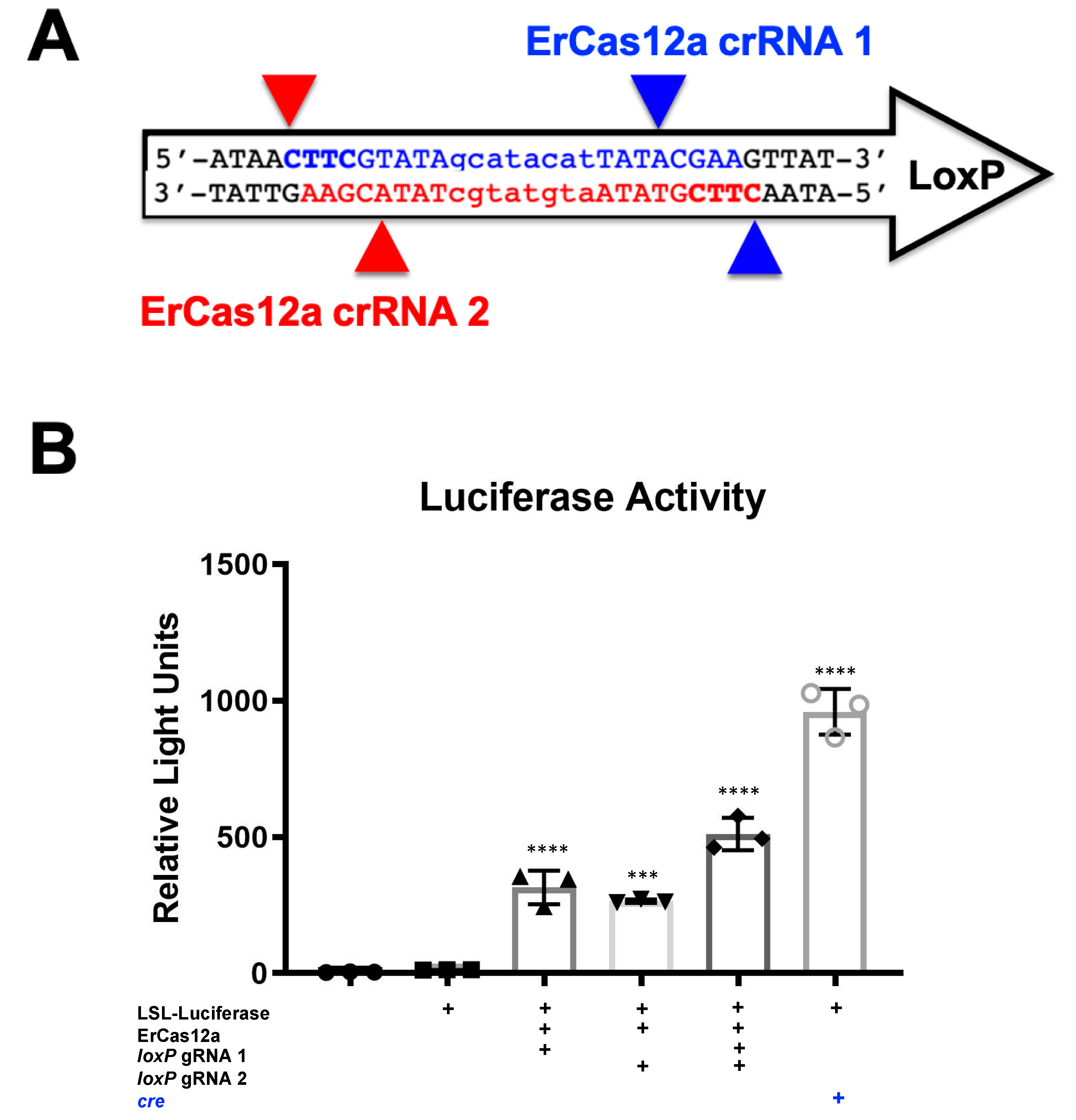
It should be noted that despite the great activity shown by SaCas9 targeting
loxP, results may vary depending on the DNA site being targeted or the CRISPR being used. This SaCas9 reporter system is highly efficient while the ErCas12a has lower activity. (
Figure 1 and
Figure 2). This can be attributed, at least partially, by differential binding and cleavage kinetics demonstrated by Cas12a type effectors compared to Cas9. It would be of interest in the future to determine if similar Cas12a effectors such as AsCas12a or LbCas12a can improve upon this foundational work with SaCas9. It is important to note that this may overestimate the activity when using a gRNA relevant to a gene therapy application or a CRISPR other than SaCas9.
This three-way reporter system can be applied to in vivo delivery of CRISPR systems to assess the tropism of the delivery system on a broad and narrow level along with a timeline of CRISPR editing. More broadly speaking, this system can be used in combination with any loxP system that relies on deletion for its activity, for which there are over 3000 mice on JAX Laboratories website related to the cre-lox system.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}