
Mechanism of Human Telomerase Reverse Transcriptase (hTERT) Regulation and Clinical Impacts in Leukemia


Abstract
1. Introduction
2. Transcriptional Regulation of TERT
2.1. Negative Transcriptional Regulators of TERT
2.2. Positive Transcriptional Regulators of TERT
2.3. Conditional Transcriptional Regulators of TERT
3. Post Transcriptional Regulation of TERT
TERT Regulation by microRNAs
4. Post Translational Regulation of TERT
5. TERT Dysregulation in Leukemias
5.1. Chronic Myeloid Leukemia (CML)
5.2. Chronic Lymphoid Leukemia (CLL)
5.3. TERT Dysregulation and Clinical Implications in Acute Leukemias
5.3.1. TERT Dysregulation and Clinical Implications in Acute Myeloid Leukemia
5.3.2. TERT Dysregulation and Clinical Implications in Acute Lymphoblastic Leukemia
6. Challenges and Opportunities in Targeting hTERT in Cancer Therapy
7. Approaches Targeting Telomerase in Preclinical and Clinical Trials
7.1. Immunotherapies
7.2. Oligonucleotide Inhibitors
7.3. Small Molecule Inhibitors
7.4. Indirect Inhibitors
7.4.1. G-Quadruplex Stabilizers
7.4.2. Nucleoside Analogues
8. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cong, Y.-S.; Wright, W.E.; Shay, J.W. Human Telomerase and Its Regulation. Microbiol. Mol. Biol. Rev. 2002, 66, 407–425. [Google Scholar] [CrossRef]
- Ly, H. Telomere Dynamics in Induced Pluripotent Stem Cells: Potentials for Human Disease Modeling. World J. Stem Cells 2011, 3, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, C.; Teixeira, M.T.; Förstemann, K.; Lingner, J. Telomerase: Biochemical Considerations for Enzyme and Substrate. Trends Biochem. Sci. 2002, 27, 572–579. [Google Scholar] [CrossRef]
- Cifuentes-Rojas, C.; Shippen, D.E. Telomerase Regulation. Mutat. Res. 2012, 730, 20–27. [Google Scholar] [CrossRef]
- Depcrynski, A.N.; Sachs, P.C.; Elmore, L.W.; Holt, S.E. Regulation of Telomerase Through Transcriptional and Posttranslational Mechanisms. In Telomeres and Telomerase in Cancer; Cancer Drug Discovery and Development; Hiyama, K., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 47–85. ISBN 978-1-60327-879-9. [Google Scholar]
- Dwyer, J.; Li, H.; Xu, D.; Liu, J.-P. Transcriptional Regulation of Telomerase Activity: Roles of the the Ets Transcription Factor Family. Ann. N. Y. Acad. Sci. 2007, 1114, 36–47. [Google Scholar] [CrossRef]
- Relitti, N.; Saraswati, A.P.; Federico, S.; Khan, T.; Brindisi, M.; Zisterer, D.; Brogi, S.; Gemma, S.; Butini, S.; Campiani, G. Telomerase-Based Cancer Therapeutics: A Review on Their Clinical Trials. Curr. Top. Med. Chem. 2020, 20, 433–457. [Google Scholar] [CrossRef]
- Bernal, A.; Tusell, L. Telomeres: Implications for Cancer Development. Int. J. Mol. Sci. 2018, 19, 294. [Google Scholar] [CrossRef] [PubMed]
- Ramlee, M.K.; Wang, J.; Toh, W.X.; Li, S. Transcription Regulation of the Human Telomerase Reverse Transcriptase (HTERT) Gene. Genes 2016, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Garbuzov, A.; Pech, M.F.; Hasegawa, K.; Sukhwani, M.; Zhang, R.J.; Orwig, K.E.; Artandi, S.E. Purification of GFRα1+ and GFRα1- Spermatogonial Stem Cells Reveals a Niche-Dependent Mechanism for Fate Determination. Stem Cell Rep. 2018, 10, 553–567. [Google Scholar] [CrossRef]
- Jaiswal, R.K.; Kumar, P.; Kumar, M.; Yadava, P.K. HTERT Promotes Tumor Progression by Enhancing TSPAN13 Expression in Osteosarcoma Cells. Mol. Carcinog. 2018, 57, 1038–1054. [Google Scholar] [CrossRef]
- Lin, S.; Nascimento, E.M.; Gajera, C.R.; Chen, L.; Neuhöfer, P.; Garbuzov, A.; Wang, S.; Artandi, S.E. Distributed Hepatocytes Expressing Telomerase Repopulate the Liver in Homeostasis and Injury. Nature 2018, 556, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Leão, R.; Apolónio, J.D.; Lee, D.; Figueiredo, A.; Tabori, U.; Castelo-Branco, P. Mechanisms of Human Telomerase Reverse Transcriptase (HTERT) Regulation: Clinical Impacts in Cancer. J. Biomed. Sci. 2018, 25, 22. [Google Scholar] [CrossRef]
- Xia, F.; Yuan, Y.; Chen, R.; Liu, P.; Zhou, H.; Wang, J. Study of telomerase activity and its related genes expression in leukemia cells. Zhonghua Xue Ye Xue Za Zhi 2001, 22, 86–89. [Google Scholar] [PubMed]
- Bruedigam, C.; Lane, S.W. Telomerase in Hematologic Malignancies. Curr. Opin. Hematol. 2016, 23, 346–353. [Google Scholar] [CrossRef]
- Davison, G.M. Telomeres and Telomerase in Leukaemia and Lymphoma. Transfus. Apher. Sci. 2007, 37, 43–47. [Google Scholar] [CrossRef]
- Keller, G.; Brassat, U.; Braig, M.; Heim, D.; Wege, H.; Brümmendorf, T.H. Telomeres and Telomerase in Chronic Myeloid Leukaemia: Impact for Pathogenesis, Disease Progression and Targeted Therapy. Hematol. Oncol. 2009, 27, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Lansdorp, P.M. Maintenance of Telomere Length in AML. Blood Adv. 2017, 1, 2467–2472. [Google Scholar] [CrossRef]
- Deville, L.; Hillion, J.; Ségal-Bendirdjian, E. Telomerase Regulation in Hematological Cancers: A Matter of Stemness? Biochim. Biophys. Acta 2009, 1792, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Pech, M.F.; Garbuzov, A.; Hasegawa, K.; Sukhwani, M.; Zhang, R.J.; Benayoun, B.A.; Brockman, S.A.; Lin, S.; Brunet, A.; Orwig, K.E.; et al. High Telomerase Is a Hallmark of Undifferentiated Spermatogonia and Is Required for Maintenance of Male Germline Stem Cells. Genes Dev. 2015, 29, 2420–2434. [Google Scholar] [CrossRef]
- Montgomery, R.K.; Carlone, D.L.; Richmond, C.A.; Farilla, L.; Kranendonk, M.E.G.; Henderson, D.E.; Baffour-Awuah, N.Y.; Ambruzs, D.M.; Fogli, L.K.; Algra, S.; et al. Mouse Telomerase Reverse Transcriptase (MTert) Expression Marks Slowly Cycling Intestinal Stem Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Chiba, K.; Johnson, J.Z.; Vogan, J.M.; Wagner, T.; Boyle, J.M.; Hockemeyer, D. Cancer-Associated TERT Promoter Mutations Abrogate Telomerase Silencing. Elife 2015, 4, e07918. [Google Scholar] [CrossRef]
- Lin, S.Y.; Elledge, S.J. Multiple Tumor Suppressor Pathways Negatively Regulate Telomerase. Cell 2003, 113, 881–889. [Google Scholar] [CrossRef]
- Parodi, S.; Ognibene, M.; Haupt, R.; Pezzolo, A. The Over-Expression of E2F3 Might Serve as Prognostic Marker for Neuroblastoma Patients with Stage 4S Disease. Diagnostics 2020, 10, 315. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Kyo, S.; Takakura, M.; Kanaya, T.; Kitagawa, Y.; Itoh, H.; Takahashi, M.; Inoue, M. Identification and Characterization of Negative Regulatory Elements of the Human Telomerase Catalytic Subunit (HTERT) Gene Promoter: Possible Role of MZF-2 in Transcriptional Repression of HTERT. Nucleic Acids Res. 2000, 28, 2557–2562. [Google Scholar] [CrossRef]
- Günes, C.; Lichtsteiner, S.; Vasserot, A.P.; Englert, C. Expression of the HTERT Gene Is Regulated at the Level of Transcriptional Initiation and Repressed by Mad1. Cancer Res. 2000, 60, 2116–2121. [Google Scholar] [PubMed]
- Gabet, A.-S.; Mortreux, F.; Charneau, P.; Riou, P.; Duc-Dodon, M.; Wu, Y.; Jeang, K.-T.; Wattel, E. Inactivation of HTERT Transcription by Tax. Oncogene 2003, 22, 3734–3741. [Google Scholar] [CrossRef]
- Kanaya, T.; Kyo, S.; Hamada, K.; Takakura, M.; Kitagawa, Y.; Harada, H.; Inoue, M. Adenoviral Expression of P53 Represses Telomerase Activity through Down-Regulation of Human Telomerase Reverse Transcriptase Transcription. Clin. Cancer Res. 2000, 6, 1239–1247. [Google Scholar] [PubMed]
- Lee, S.-H.; Kim, J.-W.; Oh, S.-H.; Kim, Y.-J.; Rho, S.-B.; Park, K.; Park, K.-L.; Lee, J.-H. IFN-Gamma/IRF-1-Induced P27kip1 down-Regulates Telomerase Activity and Human Telomerase Reverse Transcriptase Expression in Human Cervical Cancer. FEBS Lett. 2005, 579, 1027–1033. [Google Scholar] [CrossRef]
- Lacerte, A.; Korah, J.; Roy, M.; Yang, X.-J.; Lemay, S.; Lebrun, J.-J. Transforming Growth Factor-Beta Inhibits Telomerase through SMAD3 and E2F Transcription Factors. Cell. Signal. 2008, 20, 50–59. [Google Scholar] [CrossRef]
- Oh, S.; Song, Y.H.; Yim, J.; Kim, T.K. Identification of Mad as a Repressor of the Human Telomerase (HTERT) Gene. Oncogene 2000, 19, 1485–1490. [Google Scholar] [CrossRef]
- Xu, D.; Popov, N.; Hou, M.; Wang, Q.; Björkholm, M.; Gruber, A.; Menkel, A.R.; Henriksson, M. Switch from Myc/Max to Mad1/Max Binding and Decrease in Histone Acetylation at the Telomerase Reverse Transcriptase Promoter during Differentiation of HL60 Cells. Proc. Natl. Acad. Sci. USA 2001, 98, 3826–3831. [Google Scholar] [CrossRef] [PubMed]
- Sikand, K.; Kaul, D.; Varma, N. Receptor Ck-Dependent Signaling Regulates HTERT Gene Transcription. BMC Cell Biol. 2006, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Fan, S.; Meng, Q.; Schramm, L.; Wang, C.; Bouzahza, B.; Zhou, J.; Zafonte, B.; Goldberg, I.D.; Haddad, B.R.; et al. BRCA1 Inhibition of Telomerase Activity in Cultured Cells. Mol. Cell. Biol. 2003, 23, 8668–8690. [Google Scholar] [CrossRef]
- Xu, D.; Wang, Q.; Gruber, A.; Björkholm, M.; Chen, Z.; Zaid, A.; Selivanova, G.; Peterson, C.; Wiman, K.G.; Pisa, P. Downregulation of Telomerase Reverse Transcriptase MRNA Expression by Wild Type P53 in Human Tumor Cells. Oncogene 2000, 19, 5123–5133. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, D.; Li, J.; Berndt, M.C.; Liu, J.-P. Transforming Growth Factor Beta Suppresses Human Telomerase Reverse Transcriptase (HTERT) by Smad3 Interactions with c-Myc and the HTERT Gene. J. Biol. Chem. 2006, 281, 25588–25600. [Google Scholar] [CrossRef]
- Yang, H.; Kyo, S.; Takatura, M.; Sun, L. Autocrine Transforming Growth Factor Beta Suppresses Telomerase Activity and Transcription of Human Telomerase Reverse Transcriptase in Human Cancer Cells. Cell Growth Differ. 2001, 12, 119–127. [Google Scholar] [PubMed]
- Endoh, T.; Tsuji, N.; Asanuma, K.; Yagihashi, A.; Watanabe, N. Survivin Enhances Telomerase Activity via Up-Regulation of Specificity Protein 1- and c-Myc-Mediated Human Telomerase Reverse Transcriptase Gene Transcription. Exp. Cell Res. 2005, 305, 300–311. [Google Scholar] [CrossRef]
- Katzenellenbogen, R.A.; Egelkrout, E.M.; Vliet-Gregg, P.; Gewin, L.C.; Gafken, P.R.; Galloway, D.A. NFX1-123 and Poly(A) Binding Proteins Synergistically Augment Activation of Telomerase in Human Papillomavirus Type 16 E6-Expressing Cells. J. Virol. 2007, 81, 3786–3796. [Google Scholar] [CrossRef] [PubMed]
- McMurray, H.R.; McCance, D.J. Degradation of P53, Not Telomerase Activation, by E6 Is Required for Bypass of Crisis and Immortalization by Human Papillomavirus Type 16 E6/E7. J. Virol. 2004, 78, 5698–5706. [Google Scholar] [CrossRef]
- Crowe, D.L.; Nguyen, D.C. Rb and E2F-1 Regulate Telomerase Activity in Human Cancer Cells. Biochim. Biophys. Acta 2001, 1518, 1–6. [Google Scholar] [CrossRef]
- Won, J.; Yim, J.; Kim, T.K. Opposing Regulatory Roles of E2F in Human Telomerase Reverse Transcriptase (HTERT) Gene Expression in Human Tumor and Normal Somatic Cells. FASEB J. 2002, 16, 1943–1945. [Google Scholar] [CrossRef]
- Chang, J.T.-C.; Yang, H.-T.; Wang, T.-C.V.; Cheng, A.-J. Upstream Stimulatory Factor (USF) as a Transcriptional Suppressor of Human Telomerase Reverse Transcriptase (HTERT) in Oral Cancer Cells. Mol. Carcinog. 2005, 44, 183–192. [Google Scholar] [CrossRef]
- Goueli, B.S.; Janknecht, R. Regulation of Telomerase Reverse Transcriptase Gene Activity by Upstream Stimulatory Factor. Oncogene 2003, 22, 8042–8047. [Google Scholar] [CrossRef] [PubMed]
- Kyo, S.; Takakura, M.; Taira, T.; Kanaya, T.; Itoh, H.; Yutsudo, M.; Ariga, H.; Inoue, M. Sp1 Cooperates with C-Myc to Activate Transcription of the Human Telomerase Reverse Transcriptase Gene (HTERT). Nucleic Acids Res. 2000, 28, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Borah, S.; Robertson, E.S. Latency-Associated Nuclear Antigen of Kaposi’s Sarcoma-Associated Herpesvirus up-Regulates Transcription of Human Telomerase Reverse Transcriptase Promoter through Interaction with Transcription Factor Sp1. J. Virol. 2004, 78, 10348–10359. [Google Scholar] [CrossRef]
- Won, J.; Yim, J.; Kim, T.K. Sp1 and Sp3 Recruit Histone Deacetylase to Repress Transcription of Human Telomerase Reverse Transcriptase (HTERT) Promoter in Normal Human Somatic Cells. J. Biol. Chem. 2002, 277, 38230–38238. [Google Scholar] [CrossRef] [PubMed]
- Beitzinger, M.; Oswald, C.; Beinoraviciute-Kellner, R.; Stiewe, T. Regulation of Telomerase Activity by the P53 Family Member P73. Oncogene 2006, 25, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Racek, T.; Mise, N.; Li, Z.; Stoll, A.; Pützer, B.M. C-Terminal P73 Isoforms Repress Transcriptional Activity of the Human Telomerase Reverse Transcriptase (HTERT) Promoter. J. Biol. Chem. 2005, 280, 40402–40405. [Google Scholar] [CrossRef] [PubMed]
- Toh, W.H.; Kyo, S.; Sabapathy, K. Relief of P53-Mediated Telomerase Suppression by P73. J. Biol. Chem. 2005, 280, 17329–17338. [Google Scholar] [CrossRef]
- Guzman, H.; Sanders, K.; Idica, A.; Bochnakian, A.; Jury, D.; Daugaard, I.; Zisoulis, D.G.; Pedersen, I.M. MiR-128 Inhibits Telomerase Activity by Targeting TERT MRNA. Oncotarget 2018, 9, 13244–13253. [Google Scholar] [CrossRef]
- Hrdličková, R.; Nehyba, J.; Bargmann, W.; Bose, H.R. Multiple Tumor Suppressor MicroRNAs Regulate Telomerase and TCF7, an Important Transcriptional Regulator of the Wnt Pathway. PLoS ONE 2014, 9, e86990. [Google Scholar] [CrossRef]
- Jie, M.-M.; Chang, X.; Zeng, S.; Liu, C.; Liao, G.-B.; Wu, Y.-R.; Liu, C.-H.; Hu, C.-J.; Yang, S.-M.; Li, X.-Z. Diverse Regulatory Manners of Human Telomerase Reverse Transcriptase. Cell Commun. Signal. 2019, 17, 63. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Xu, D. Telomerase Reverse Transcriptase (TERT) in Action: Cross-Talking with Epigenetics. Int. J. Mol. Sci. 2019, 20, 3338. [Google Scholar] [CrossRef]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms Underlying the Activation of TERT Transcription and Telomerase Activity in Human Cancer: Old Actors and New Players. Oncogene 2019, 38, 6172–6183. [Google Scholar] [CrossRef] [PubMed]
- Si, W.; Shen, J.; Zheng, H.; Fan, W. The Role and Mechanisms of Action of MicroRNAs in Cancer Drug Resistance. Clin. Epigenet. 2019, 11, 25. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of MicroRNA Biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The Role of MicroRNAs in Human Cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Bai, L.; Wang, H.; Wang, A.-H.; Zhang, L.-Y.; Bai, J. MicroRNA-532 and MicroRNA-3064 Inhibit Cell Proliferation and Invasion by Acting as Direct Regulators of Human Telomerase Reverse Transcriptase in Ovarian Cancer. PLoS ONE 2017, 12, e0173912. [Google Scholar] [CrossRef]
- Farooqi, A.A.; Mansoor, Q.; Alaaeddine, N.; Xu, B. MicroRNA Regulation of Telomerase Reverse Transcriptase (TERT): Micro Machines Pull Strings of Papier-Mâché Puppets. Int. J. Mol. Sci. 2018, 19, 1051. [Google Scholar] [CrossRef]
- Gagnon, J.D.; Kageyama, R.; Shehata, H.M.; Fassett, M.S.; Mar, D.J.; Wigton, E.J.; Johansson, K.; Litterman, A.J.; Odorizzi, P.; Simeonov, D.; et al. MiR-15/16 Restrain Memory T Cell Differentiation, Cell Cycle, and Survival. Cell Rep. 2019, 28, 2169–2181.e4. [Google Scholar] [CrossRef]
- Manasa, V.G.; Kannan, S. Impact of MicroRNA Dynamics on Cancer Hallmarks: An Oral Cancer Scenario. Tumour Biol. 2017, 39, 1010428317695920. [Google Scholar] [CrossRef]
- Hou, X.-S.; Han, C.-Q.; Zhang, W. MiR-1182 Inhibited Metastasis and Proliferation of Ovarian Cancer by Targeting HTERT. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 1622–1628. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zhai, Y.-X.; Liu, H.-Q.; Shi, Y.-A.; Li, X.-B. MicroRNA-491-5p Suppresses Cervical Cancer Cell Growth by Targeting HTERT. Oncol. Rep. 2015, 34, 979–986. [Google Scholar] [CrossRef]
- Huang, Y. Plk1 Upregulates Telomerase Activity by Affecting HTERT Stability. JBC 2015, 290, 18865–18873. [Google Scholar] [CrossRef] [PubMed]
- Yalçin, Z.; Selenz, C.; Jacobs, J.J.L. Ubiquitination and SUMOylation in Telomere Maintenance and Dysfunction. Front. Genet. 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lanna, A.; Coutavas, E.; Levati, L.; Rustin, M.H.A.; Henson, S.M.; Arne, N. IFN-α Inhibits Telomerase in Human CD8 + T Cells by Both HTERT Downregulation and Induction of P38 MAPK Signaling. J. Immunol. 2020, 191, 3744–3752. [Google Scholar] [CrossRef]
- Nalobin, D.S.; Galiakberova, A.A.; Alipkina, S.I.; Glukhov, A.I. Regulation of Telomerase Activity. Biol. Bull. Rev. 2018, 8, 142–154. [Google Scholar] [CrossRef]
- Macneil, D.E.; Bensoussan, H.J.; Autexier, C. Telomerase Regulation from Beginning to the End. Genes 2016, 7, 64. [Google Scholar] [CrossRef]
- Singh, R.K.; Kumar, S.; Gautam, P.K.; Tomar, M.S.; Verma, P.K.; Singh, S.P.; Kumar, S.; Acharya, A. Protein Kinase C- α and the Regulation of Diverse Cell Responses. Biomol. Concepts 2017, 8, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.; Zhang, S.; Chen, B.P.C. DNA–Dependent Protein Kinase in Telomere Maintenance and Protection. Cell. Mol. Biol. Lett. 2020, 25, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Baloghova, N.; Lidak, T.; Cermak, L. TGF-β, and Notch Signaling Pathways and Their Roles in Mouse Development and Homeostasis. Genes 2019, 10, 815. [Google Scholar] [CrossRef] [PubMed]
- Sluimer, J.; Distel, B. Regulating the Human HECT E3 Ligases. Cell. Mol. Life Sci. 2018, 75, 3121–3141. [Google Scholar] [CrossRef] [PubMed]
- Kim, J. Alternative Splicing and Extra-Telomeric Role of Telomerase Reverse Transcriptase Isoforms in Human Embryonic Stem Cells. Masters Thesis, Western University, London, ON, Canada, 2017. [Google Scholar]
- Pourbagheri-Sigaroodi, A.; Bashash, D.; Safaroghli-Azar, A.; Farshi-Paraasghari, M.; Momeny, M.; Mansoor, F.N.; Ghaffari, S.H. Contributory Role of MicroRNAs in Anti-Cancer Effects of Small Molecule Inhibitor of Telomerase (BIBR1532) on Acute Promyelocytic Leukemia Cell Line. Eur. J. Pharmacol. 2019, 846, 49–62. [Google Scholar] [CrossRef]
- Liu, X.; Zou, L.; Zhu, L.; Zhang, H.; Du, C.; Li, Z.; Gao, C.; Zhao, X.; Bao, S.; Zheng, H. MiRNA Mediated Up-Regulation of Cochaperone P23 Acts as an Anti-Apoptotic Factor in Childhood Acute Lymphoblastic Leukemia. Leuk. Res. 2012, 36, 1098–1104. [Google Scholar] [CrossRef]
- Bhatia, S.; Kaul, D.; Varma, N. Functional Genomics of Tumor Suppressor MiR-196b in T-Cell Acute Lymphoblastic Leukemia. Mol. Cell Biochem. 2011, 346, 103–116. [Google Scholar] [CrossRef]
- El Hajj, J.; Nguyen, E.; Liu, Q.; Bouyer, C.; Adriaenssens, E.; Hilal, G.; Ségal-Bendirdjian, E. Telomerase Regulation by the Long Non-Coding RNA H19 in Human Acute Promyelocytic Leukemia Cells. Mol. Cancer 2018, 17, 85. [Google Scholar] [CrossRef]
- Abdelrahman, A.H.; Eid, M.M.; Hassan, M.; Eid, O.M.; AbdelKader, R.M.A.; AlAzhary, N.M.; Shahin, R.Y.; Sallam, M.T. Telomerase Reverse Transcriptase Gene Amplification in Hematological Malignancies. Egypt J. Med. Hum. Genet. 2019, 20, 1–9. [Google Scholar] [CrossRef]
- Ghaffari, S.H.; Shayan-Asl, N.; Jamialahmadi, A.H.; Alimoghaddam, K.; Ghavamzadeh, A. Telomerase Activity and Telomere Length in Patients with Acute Promyelocytic Leukemia: Indicative of Proliferative Activity, Disease Progression, and Overall Survival. Ann. Oncol. 2008, 19, 1927–1934. [Google Scholar] [CrossRef]
- Ishida, H.; Iguchi, A.; Aoe, M.; Takahashi, T.; Tamefusa, K.; Kanamitsu, K.; Fujiwara, K.; Washio, K.; Matsubara, T.; Tsukahara, H.; et al. Panel-Based next-Generation Sequencing Identifies Prognostic and Actionable Genes in Childhood Acute Lymphoblastic Leukemia and Is Suitable for Clinical Sequencing. Ann. Hematol. 2019, 98, 657–668. [Google Scholar] [CrossRef]
- Alsultan, A.; Essa, M.; Aljefri, A.; Ayas, M.; Alharbi, M.; Alkhayat, N.; Al-Anzi, F.; Yassin, F.; Alkasim, F.; Alharbi, Q.; et al. Frequency of Pathogenic/Likely Pathogenic Germline Variants in Cancer-Related Genes among Children with Acute Leukemia in Saudi Arabia. Pediatr. Blood Cancer 2020, 67, e28340. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Dueñas, C.; Barajas-Diego, M.; Romero-Camarero, I.; González-Herrero, I.; Flores, T.; Sánchez-García, I. Essential Role for Telomerase in Chronic Myeloid Leukemia Induced by BCR-ABL in Mice. Oncotarget 2012, 3, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Röth, A.; Vercauteren, S.; Sutherland, H.J.; Lansdorp, P.M. Telomerase Is Limiting the Growth of Acute Myeloid Leukemia Cells. Leukemia 2003, 17, 2410–2417. [Google Scholar] [CrossRef] [PubMed]
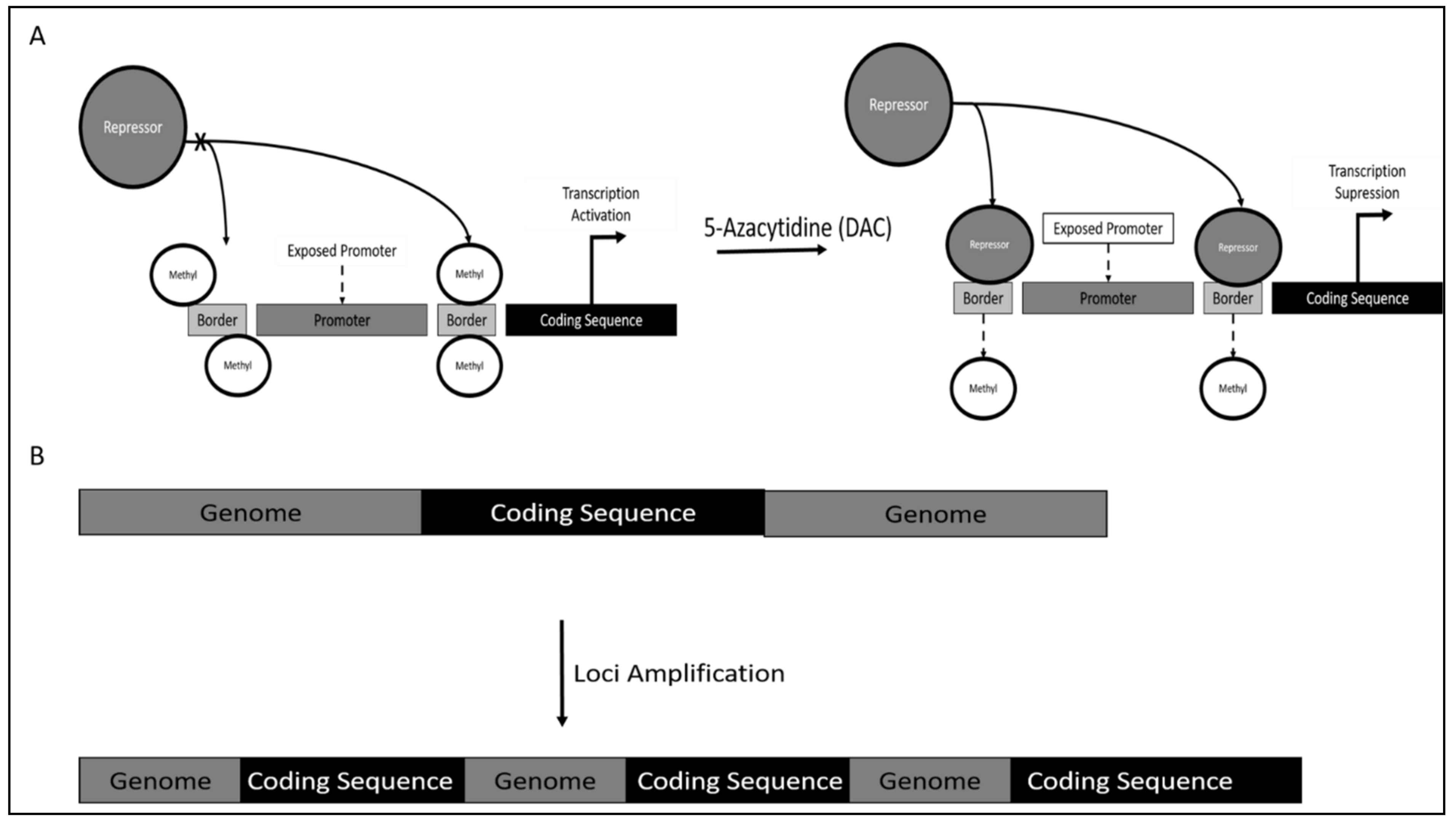
- Grandjenette, C.; Schnekenburger, M.; Karius, T.; Ghelfi, J.; Gaigneaux, A.; Henry, E.; Dicato, M.; Diederich, M. 5-Aza-2′-Deoxycytidine-Mediated c-Myc Down-Regulation Triggers Telomere-Dependent Senescence by Regulating Human Telomerase Reverse Transcriptase in Chronic Myeloid Leukemia. Neoplasia 2014, 16, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Mohamad Ashari, Z.S.; Sulong, S.; Hassan, R.; Husin, A.; Sim, G.A.; Wahid, S.F.A. Low Level of TERC Gene Amplification between Chronic Myeloid Leukaemia Patients Resistant and Respond to Imatinib Mesylate Treatment. Asian Pac. J. Cancer Prev. 2014, 15, 1863–1869. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.H.; Zhang, Y.; Tan, W.H.; Chng, W.J.; Li, B.; Wang, X. Regulation of HTERT by BCR-ABL at Multiple Levels in K562 Cells. BMC Cancer 2011, 11, 512. [Google Scholar] [CrossRef] [PubMed]
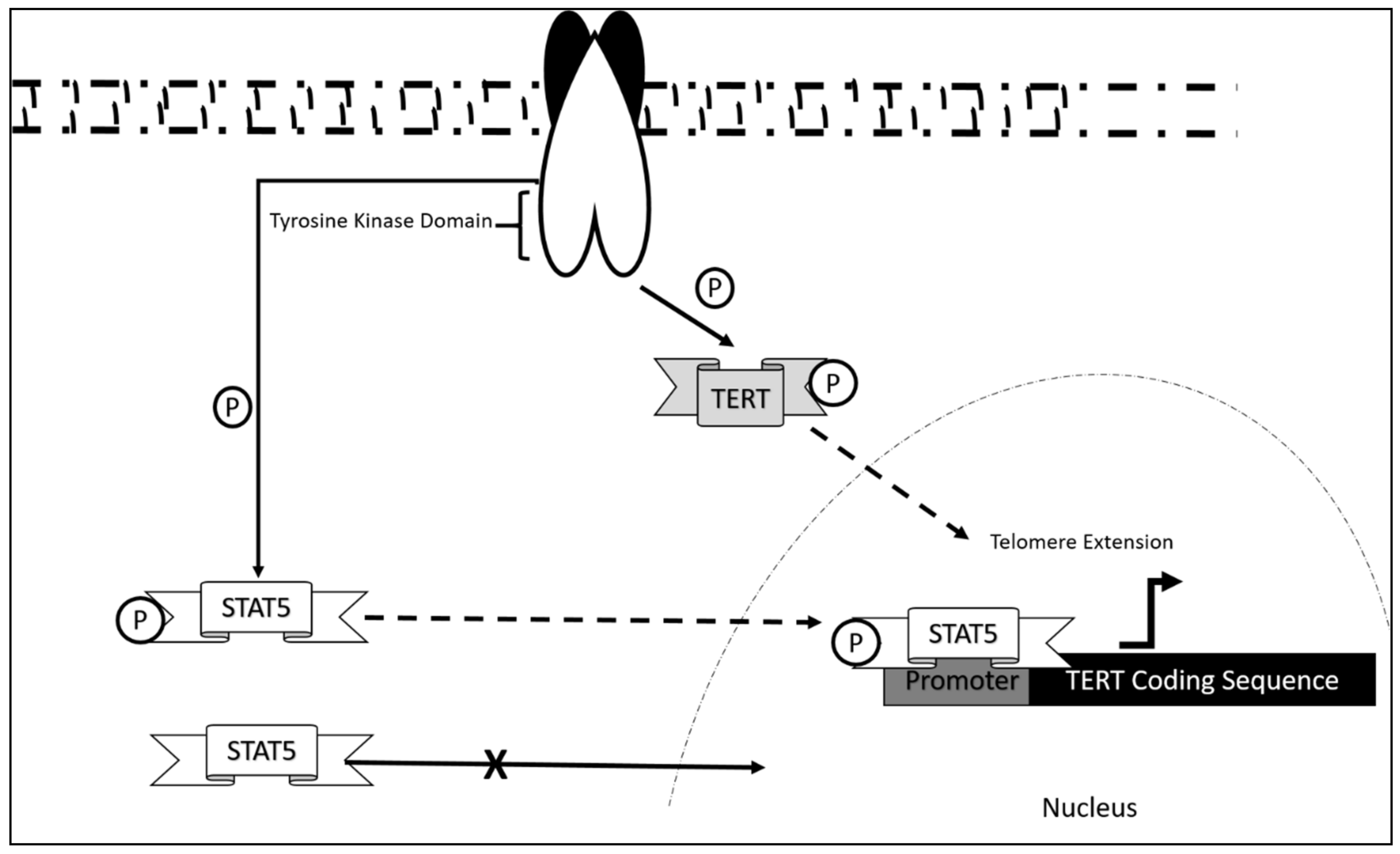
- Rondanin, R.; Simoni, D.; Romagnoli, R.; Baruchello, R.; Marchetti, P.; Costantini, C.; Fochi, S.; Padroni, G.; Grimaudo, S.; Pipitone, R.M. Inhibition of Activated STAT5 in Bcr/Abl Expressing Leukemia Cells with New Pimozide Derivatives. Bioorg. Med. Chem. Lett. 2014, 24, 4568–4574. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K. Activation of STAT5 Confers Imatinib Resistance on Leukemic Cells. Ph.D. Thesis, Department of Hematology, Tokyo Women’s Medical University, Tokyo, Japan, 2011. [Google Scholar]
- Yamada, O.; Ozaki, K.; Furukawa, T.; Machida, M.; Wang, Y.-H.; Motoji, T.; Mitsuishi, T.; Akiyama, M.; Yamada, H.; Kawauchi, K.; et al. Activation of STAT5 Confers Imatinib Resistance on Leukemic Cells through the Transcription of TERT and MDR1. Cell. Signal. 2011, 23, 1119–1127. [Google Scholar] [CrossRef]
- Grandjenette, C.; Schnekenburger, M.; Gaigneaux, A.; Gérard, D.; Christov, C.; Mazumder, A.; Dicato, M.; Diederich, M. Human Telomerase Reverse Transcriptase Depletion Potentiates the Growth-Inhibitory Activity of Imatinib in Chronic Myeloid Leukemia Stem Cells. Cancer Lett. 2020, 469, 468–480. [Google Scholar] [CrossRef]
- Akiyama, M.; Yamada, O.; Akita, S.; Urashima, M.; Horiguchi-Yamada, J.; Ohno, T.; Mizoguchi, H.; Eto, Y.; Yamada, H. Ectopic Expression of C- Myc Fails to Overcome Downregulation of Telomerase Activity Induced by Herbimycin A, but Ectopic HTERT Expression Overcomes It. Leukemia 2000, 14, 1260–1265. [Google Scholar] [CrossRef][Green Version]
- Celeghin, A.; Giunco, S.; Freguja, R.; Zangrossi, M.; Nalio, S.; Dolcetti, R.; Rossi, A.D. Short-Term Inhibition of TERT Induces Telomere Length-Independent Cell Cycle Arrest and Apoptotic Response in EBV-Immortalized and Transformed B Cells. Cell Death Dis. 2016, 7, e2562. [Google Scholar] [CrossRef]
- Augereau, A.; T’kint de Roodenbeke, C.; Simonet, T.; Bauwens, S.; Horard, B.; Callanan, M.; Leroux, D.; Jallades, L.; Salles, G.; Gilson, E.; et al. Telomeric Damage in Early Stage of Chronic Lymphocytic Leukemia Correlates with Shelterin Dysregulation. Blood 2011, 118, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Hoxha, M.; Fabris, S.; Agnelli, L.; Bollati, V.; Cutrona, G.; Matis, S.; Recchia, A.G.; Gentile, M.; Cortelezzi, A.; Morabito, F.; et al. Relevance of Telomere/Telomerase System Impairment in Early Stage Chronic Lymphocytic Leukemia. Genes Chromosomes Cancer 2014, 53, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Katsumi, S.; Kawauchi, K.; Ozaki, K.; Shimizu, S.; Kimura, T.; Motoji, T.; Yamada, O. Analysis of Molecular Mechanism Involved in Development of Acute Myeloid Leukemia. Gan Kagaku Ryoho Cancer Chemother. 2013, 40, 471–477. [Google Scholar]
- Schafranek, L.; Nievergall, E.; Powell, J.A.; Hiwase, D.K.; Leclercq, T.; Hughes, T.P.; White, D.L. Sustained Inhibition of STAT5, but Not JAK2, Is Essential for TKI-Induced Cell Death in Chronic Myeloid Leukemia. Leukemia 2015, 29, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Chiba, K.; Lorbeer, F.K.; Shain, A.H.; McSwiggen, D.T.; Schruf, E.; Oh, A.; Ryu, J.; Darzacq, X.; Bastian, B.C.; Hockemeyer, D. Mutations in the Promoter of the Telomerase Gene TERT Contribute to Tumorigenesis by a Two-Step Mechanism. Science 2017, 357, 1416–1420. [Google Scholar] [CrossRef] [PubMed]
- Fonseka, L.N.; Tirado, C.A. Telomerase in Acute Myeloid Leuk Emia: A Molecular Update on Diagnosis, Prognosis, and Treatment. J. Assoc. Genet. Technol. 2016, 42, 105–110. [Google Scholar] [PubMed]
- Shahjahani, M.; Abroun, A.; Saki, N.; Mohammadi, B.; Mohammad, S.; Rezaeeyan, H. STAT5: From Pathogenesis Mechanism to Therapeutic Approach in Acute Leukemia. Lab. Med. 2019, 51, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Ghamlouch, H.; Nguyen-Khac, F.; Bernard, O.A. Chronic Lymphocytic Leukaemia Genomics and the Precision Medicine Era. Br. J. Haematol. 2017, 178, 852–870. [Google Scholar] [CrossRef] [PubMed]
- Wysoczanska, B.; Dratwa, M.; Gebura, K.; Mizgala, J.; Mazur, G.; Wrobel, T.; Bogunia-Kubik, K. Variability within the Human TERT Gene, Telomere Length and Predisposition to Chronic Lymphocytic Leukemia. OncoTargets Ther. 2019, 12, 4309–4320. [Google Scholar] [CrossRef]
- Ojha, J.; Codd, V.; Nelson, C.P.; Samani, N.J.; Smirnov, I.V.; Madsen, N.R.; Hansen, H.M.; de Smith, A.J.; Bracci, P.M.; Wiencke, J.K. Genetic Variation Associated with Longer Telomere Length Increases Risk of Chronic Lymphocytic Leukemia. Cancer Epidemiol. Prev. Biomark. 2016, 25, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.M.; Beggs, A.D.; Carvajal-Carmona, L.; Farrington, S.; Tenesa, A.; Walker, M.; Howarth, K.; Ballereau, S.; Hodgson, S.V.; Zauber, A.; et al. TERC Polymorphisms Are Associated Both with Susceptibility to Colorectal Cancer and with Longer Telomeres. Gut 2012, 61, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Küppers, R.; Dalla-Favera, R. Mechanisms of Chromosomal Translocations in B Cell Lymphomas. Oncogene 2001, 20, 5580–5594. [Google Scholar] [CrossRef] [PubMed]
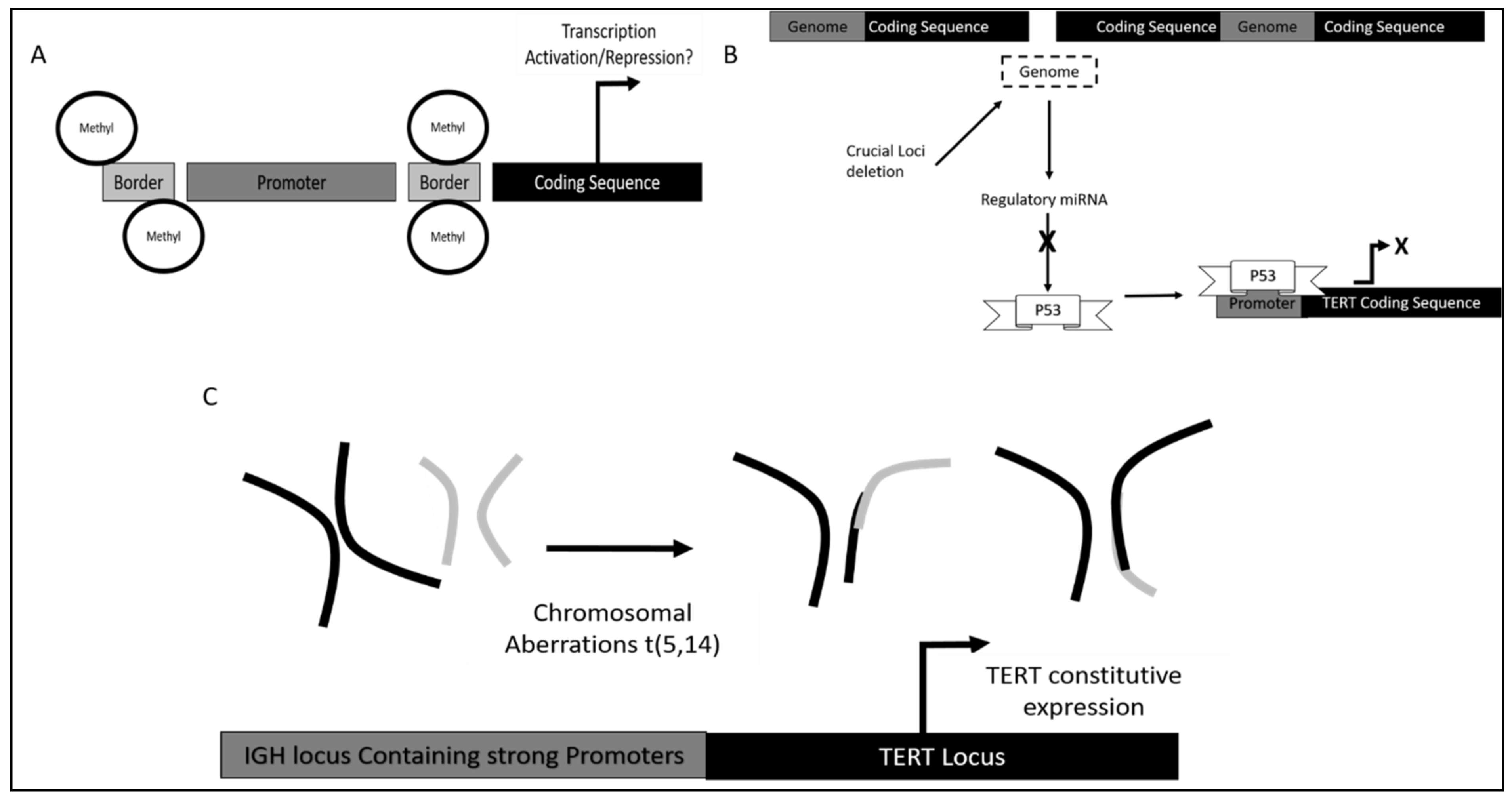
- Nagel, I.; Szczepanowski, M.; Martín-Subero, J.I.; Harder, L.; Akasaka, T.; Ammerpohl, O.; Callet-Bauchu, E.; Gascoyne, R.D.; Gesk, S.; Horsman, D.; et al. Deregulation of the Telomerase Reverse Transcriptase (TERT) Gene by Chromosomal Translocations in B-Cell Malignancies. Blood 2010, 116, 1317–1320. [Google Scholar] [CrossRef]
- Andreani, G.; Carrà, G.; Lingua, M.F.; Maffeo, B.; Brancaccio, M.; Taulli, R.; Morotti, A. Tumor Suppressors in Chronic Lymphocytic Leukemia: From Lost Partners to Active Targets. Cancers 2020, 12, 629. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, E.; Bojnik, E.; Trentin, L.; Bonaldi, L.; Del Bianco, P.; Frezzato, F.; Visentin, A.; Facco, M.; Semenzato, G.; De Rossi, A. Role of MiR-15a/MiR-16-1 and the TP53 Axis in Regulating Telomerase Expression in Chronic Lymphocytic Leukemia. Haematologica 2017, 102, e253–e256. [Google Scholar] [CrossRef]
- Shawi, M. Telomerase and Telomere Regulation by Associated Proteins and in Primary Malignant Lymphocytes. Ph.D. Thesis, McGill University, Montreal, QC, Canada, 2012. [Google Scholar]
- Tefferi, A.; Lasho, T.L.; Begna, K.H.; Patnaik, M.M.; Zblewski, D.L.; Finke, C.M.; Laborde, R.R.; Wassie, E.; Schimek, L.; Hanson, C.A.; et al. A Pilot Study of the Telomerase Inhibitor Imetelstat for Myelofibrosis. N. Engl. J. Med. 2015, 373, 908–919. [Google Scholar] [CrossRef]
- Wang, S.; Pike, A.M.; Lee, S.S.; Strong, M.A.; Connelly, C.J.; Greider, C.W. BRD4 Inhibitors Block Telomere Elongation. Nucleic Acids Res. 2017, 45, 8403–8410. [Google Scholar] [CrossRef]
- Hoffbrand, V.A.; Moss, P.A.H. Hoffbrand’s Essential Hematology, 7th ed.; Wiley Blackwell: Noida, India, October 2015. [Google Scholar]
- Lin, S.; Wei, J.; Wunderlich, M.; Chou, F.S.; Mulloy, J.C. Immortalization of Human AE Pre-Leukemia Cells by HTERT Allows Leukemic Transformation. Oncotarget 2016, 7, 55939–55950. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eid, M.M.; Helmy, N.A.; Omar, I.M.; Mohamed, A.A.; El Sewefy, D.; Fadel, I.M.; Helal, R.A. Clinical Significance of Telomerase Genes (HTERC and HTERT) Amplification in Patients with Acute Myeloid Leukemia. Gulf J. Oncol. 2013, 1, 51–60. [Google Scholar]
- Nowak, T.; Januszkiewicz, D.; Zawada, M.; Pernak, M.; Lewandowski, K.; Rembowska, J.; Nowicka, K.; Mankowski, P.; Nowak, J. Amplification of HTERT and HTERC Genes in Leukemic Cells with High Expression and Activity of Telomerase. Oncol. Rep. 2006, 16, 301–305. [Google Scholar] [CrossRef][Green Version]
- Syibli, O.A.; Ashari, Z.S.M.; Adam, N.A.M.; Ankathil, R.; Hassan, R.; Sulong, S. Detection of TERT Amplification in Cancer Cells Using Fluorescence in Situ Hybridization and Quantitative Real Time PCR Assays. Int. Med. J. 2013, 20, 272–276. [Google Scholar]
- Spencer, D.H.; Zhang, B.; Pfeifer, J. Single Nucleotide Variant Detection Using Next Generation Sequencing. In Clinical Genomics; Academic Press: Cambridge, MA, USA, 2015; ISBN 978-0-12-405173-7. [Google Scholar]
- Mosrati, M.A.; Willander, K.; Falk, I.J.; Hermanson, M.; Höglund, M.; Stockelberg, D.; Wei, Y.; Lotfi, K.; Söderkvist, P. Association between TERT Promoter Polymorphisms and Acute Myeloid Leukemia Risk and Prognosis. Oncotarget 2015, 6, 25109. [Google Scholar] [CrossRef] [PubMed]
- Aref, S.; El-Ghonemy, M.S.; Abouzeid, T.E.; El-Sabbagh, A.M.; El-Baiomy, M.A. Telomerase Reverse Transcriptase (TERT) A1062T Mutation as a Prognostic Factor in Egyptian Patients with Acute Myeloid Leukemia (AML). Med. Oncol. 2014, 31, 158. [Google Scholar] [CrossRef] [PubMed]
- Both, A.; Krauter, J.; Damm, F.; Thol, F.; Göhring, G.; Heuser, M.; Ottmann, O.; Lübbert, M.; Wattad, M.; Kanz, L.; et al. The Hypomorphic TERT A1062T Variant Is Associated with Increased Treatment-Related Toxicity in Acute Myeloid Leukemia. Ann. Hematol. 2017, 96, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Calado, R.T.; Regal, J.A.; Hills, M.; Yewdell, W.T.; Dalmazzo, L.F.; Zago, M.A.; Lansdorp, P.M.; Hogge, D.; Chanock, S.J.; Estey, E.H.; et al. Constitutional Hypomorphic Telomerase Mutations in Patients with Acute Myeloid Leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.; Both, A.; Damm, F.; Thol, F.; Göhring, G.; Heuser, M.; Ottmann, O.G.; Lübbert, M.; Wattad, M.; Kanz, L.; et al. Clinical Impact of TERT A1062T Mutations in Younger Patients with Acute Myeloblastic Leukemia. Blood 2012, 120, 1381. [Google Scholar] [CrossRef]
- Lee, D.D.; Leão, R.; Komosa, M.; Gallo, M.; Zhang, C.H.; Lipman, T.; Remke, M.; Heidari, A.; Nunes, N.M.; Apolónio, J.D.; et al. DNA Hypermethylation within TERT Promoter Upregulates TERT Expression in Cancer. J. Clin. Investig. 2019, 129, 223–229. [Google Scholar] [CrossRef]
- Zhao, X.; Tian, X.; Kajigaya, S.; Cantilena, C.R.; Strickland, S.; Savani, B.N.; Mohan, S.; Feng, X.; Keyvanfar, K.; Dunavin, N.; et al. Epigenetic Landscape of the TERT Promoter: A Potential Biomarker for High Risk AML/MDS. Br. J. Haematol. 2016, 175, 427–439. [Google Scholar] [CrossRef]
- Staudt, D.; Murray, H.C.; McLachlan, T.; Alvaro, F.; Enjeti, A.K.; Verrills, N.M.; Dun, M.D. Targeting Oncogenic Signaling in Mutant FLT3 Acute Myeloid Leukemia: The Path to Least Resistance. Int. J. Mol. Sci. 2018, 19, 3198. [Google Scholar] [CrossRef]
- Zhang, X.; Li, B.; Yu, J.; Dahlström, J.; Tran, A.N.; Björkhom, M.; Xu, D. MYC-Dependent Downregulation of Telomerase by FLT3 Inhibitors Is Required for Their Therapeutic Efficacy on Acute Myeloid Leukemia. Ann. Hematol. 2018, 97, 63–72. [Google Scholar] [CrossRef]
- Osterwald, S.; Deeg, K.I.; Chung, I.; Parisotto, D.; Wörz, S.; Rohr, K.; Erfle, H.; Rippe, K. PML Induces Compaction, TRF2 Depletion and DNA Damage Signaling at Telomeres and Promotes Their Alternative Lengthening. J. Cell Sci. 2015, 128, 1887–1900. [Google Scholar] [CrossRef]
- Garsuault, D.; Bouyer, C.; Nguyen, E.; Kandhari, R.; Prochazkova-Carlotti, M.; Chevret, E.; Forgez, P.; Ségal-Bendirdjian, E. Complex Context Relationships between DNA Methylation and Accessibility, Histone Marks, and HTERT Gene Expression in Acute Promyelocytic Leukemia Cells: Perspectives for All-Trans Retinoic Acid in Cancer Therapy. Mol. Oncol. 2020, 14, 1310–1326. [Google Scholar] [CrossRef]
- Hu, Q.; Chen, X.; Liu, S.; Wen, R.; Yuan, X.; Xu, D.; Liu, G.; Wen, F. Methylation of CDKN2B CpG Islands Is Associated with Upregulated Telomerase Activity in Children with Acute Lymphoblastic Leukemia. Oncol. Lett. 2017, 13, 2115–2120. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, X.; Li, B.; de Jonge, N.; Björkholm, M.; Xu, D. The DNA Methylation Inhibitor Induces Telomere Dysfunction and Apoptosis of Leukemia Cells That Is Attenuated by Telomerase Over-Expression. Oncotarget 2015, 6, 4888. [Google Scholar] [CrossRef] [PubMed]
- Pettigrew, K.A.; Armstrong, R.N.; Colyer, H.A.A.; Zhang, S.D.; Rea, I.M.; Jones, R.E.; Baird, D.M.; Mills, K.I. Differential TERT Promoter Methylation and Response to 5-Aza-2′-Deoxycytidine in Acute Myeloid Leukemia Cell Lines: TERT Expression, Telomerase Activity, Telomere Length, and Cell Death. Genes Chromosomes Cancer 2012, 51, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Meng, M.; Zheng, N.; Cao, Z.; Yang, P.; Xi, X.; Zhou, Q. Targeting of Multiple Senescence-Promoting Genes and Signaling Pathways by Triptonide Induces Complete Senescence of Acute Myeloid Leukemia Cells. Biochem. Pharmacol. 2017, 126, 34–51. [Google Scholar] [CrossRef]
- Sandri, S.; De Sanctis, F.; Lamolinara, A.; Boschi, F.; Poffe, O.; Trovato, R.; Fiore, A.; Sartori, S.; Sbarbati, A.; Bondanza, A.; et al. Effective Control of Acute Myeloid Leukaemia and Acute Lymphoblastic Leukaemia Progression by Telomerase Specific Adoptive T-Cell Therapy. Oncotarget 2017, 8, 86987. [Google Scholar] [CrossRef] [PubMed]
- Khoury, H.J.; Collins, R.H.; Blum, W.; Stiff, P.S.; Elias, L.; Lebkowski, J.S.; Reddy, A.; Nishimoto, K.P.; Sen, D.; Wirth, E.D.; et al. Immune Responses and Long-Term Disease Recurrence Status after Telomerase-Based Dendritic Cell Immunotherapy in Patients with Acute Myeloid Leukemia. Cancer 2017, 123, 3061–3072. [Google Scholar] [CrossRef] [PubMed]
- Amin, D.G.; Anis, S.K.; Younan, S.; Al Gamal, R.A. Human Telomerase Reverse Transcriptase (HTERT) Expression and Telomerase Activity in Acute and Chronic Lymphocytic Leukemia Patients. UHOD Uluslar. Hematol. Onkol. Derg. 2018, 28, 61–68. [Google Scholar] [CrossRef]
- Di Maio, M. Single nucleotide polymorphisms in cancer research and treatment. In Oncogenomics: From Basic Research to Precision Medicine; Academic Press: Cambridge, MA, USA, 2018; pp. 233–242. ISBN 978-0-12-811785-9. [Google Scholar]
- Sheng, X.; Tong, N.; Tao, G.; Luo, D.; Wang, M.; Fang, Y.; Li, J.; Xu, M.; Zhang, Z.; Wu, D. TERT Polymorphisms Modify the Risk of Acute Lymphoblastic Leukemia in Chinese Children. Carcinogenesis 2013, 34, 228–235. [Google Scholar] [CrossRef]
- Eskandari, E.; Hashemi, M.; Naderi, M.; Bahari, G.; Safdari, V.; Taheri, M. Leukocyte Telomere Length Shortening, HTERT Genetic Polymorphisms and Risk of Childhood Acute Lymphoblastic Leukemia. Asian Pac. J. Cancer Prev. 2018, 19, 1515. [Google Scholar] [CrossRef] [PubMed]
- Borssén, M.; Cullman, I.; Norén-Nyström, U.; Sundström, C.; Porwit, A.; Forestier, E.; Roos, G. HTERT Promoter Methylation and Telomere Length in Childhood Acute Lymphoblastic Leukemia-Associations with Immunophenotype and Cytogenetic Subgroup. Exp. Hematol. 2011, 39, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Bashash, D.; Zareii, M.; Safaroghli-Azar, A.; Omrani, M.D.; Ghaffari, S.H. Inhibition of Telomerase Using BIBR1532 Enhances Doxorubicin-Induced Apoptosis in Pre-B Acute Lymphoblastic Leukemia Cells. Hematology 2017, 22, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Guterres, A.N.; Villanueva, J. Targeting Telomerase for Cancer Therapy. Oncogene 2020, 39, 5811–5824. [Google Scholar] [CrossRef]
- Cheng, D.; Zhao, Y.; Zhang, F.; Zhang, J.; Wang, S.; Zhu, J. Engineering a Humanized Telomerase Reverse Transcriptase Gene in Mouse Embryonic Stem Cells. Sci. Rep. 2019, 9, 9683. [Google Scholar] [CrossRef]
- Nguyen, T.H.D.; Tam, J.; Wu, R.A.; Greber, B.J.; Toso, D.; Nogales, E.; Collins, K. Cryo-EM Structure of Substrate-Bound Human Telomerase Holoenzyme. Nature 2018, 557, 190–195. [Google Scholar] [CrossRef]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of Telomeres and Telomerase in Cancer, and Advances in Telomerase-Targeted Therapies. Genome Med. 2016, 8, 1–18. [Google Scholar] [CrossRef]
- Lilleby, W.; Gaudernack, G.; Brunsvig, P.F.; Vlatkovic, L.; Schulz, M.; Mills, K.; Hole, K.H.; Inderberg, E.M. Phase I/IIa Clinical Trial of a Novel HTERT Peptide Vaccine in Men with Metastatic Hormone-Naive Prostate Cancer. Cancer Immunol. Immunother. 2017, 66, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Middleton, G.; Silcocks, P.; Cox, T.; Valle, J.; Wadsley, J.; Propper, D.; Coxon, F.; Ross, P.; Madhusudan, S.; Roques, T.; et al. Gemcitabine and Capecitabine with or without Telomerase Peptide Vaccine GV1001 in Patients with Locally Advanced or Metastatic Pancreatic Cancer (TeloVac): An Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2014, 15, 829–840. [Google Scholar] [CrossRef]
- Duperret, E.K.; Wise, M.C.; Trautz, A.; Villarreal, D.O.; Ferraro, B.; Walters, J.; Yan, J.; Khan, A.; Masteller, E.; Humeau, L.; et al. Synergy of Immune Checkpoint Blockade with a Novel Synthetic Consensus DNA Vaccine Targeting TERT. Mol. Ther. 2018, 26, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Ugel, S.; Scarselli, E.; Iezzi, M.; Mennuni, C.; Pannellini, T.; Calvaruso, F.; Cipriani, B.; De Palma, R.; Ricci-Vitiani, L.; Peranzoni, E.; et al. Autoimmune B-Cell Lymphopenia after Successful Adoptive Therapy with Telomerase-Specific T Lymphocytes. Blood 2010, 115, 1374–1384. [Google Scholar] [CrossRef] [PubMed]
- Sandri, S.; Bobisse, S.; Moxley, K.; Lamolinara, A.; De Sanctis, F.; Boschi, F.; Sbarbati, A.; Fracasso, G.; Ferrarini, G.; Hendriks, R.W.; et al. Feasibility of Telomerase-Specific Adoptive T-Cell Therapy for B-Cell Chronic Lymphocytic Leukemia and Solid Malignancies. Cancer Res. 2016, 76, 2540–2551. [Google Scholar] [CrossRef] [PubMed]
- Kanaya, N.; Kuroda, S.; Morihiro, T.; Kakiuchi, Y.; Kubota, T.; Kakiuchi, S.; Nishizaki, M.; Urata, Y.; Tazawa, H.; Kagawa, S.; et al. Abstract 2744: Telomelysin-Induced Immunogenic Cell Death Synergizes with Anti-PD-1 Antibody in Non-Immunogenic Gastrointestinal Tumors. Cancer Res. 2018, 78, 2744. [Google Scholar] [CrossRef]
- Thompson, P.A.; Drissi, R.; Muscal, J.A.; Panditharatna, E.; Fouladi, M.; Ingle, A.M.; Ahern, C.H.; Reid, J.M.; Lin, T.; Weigel, B.J.; et al. A Phase I Trial of Imetelstat in Children with Refractory or Recurrent Solid Tumors: A Children’s Oncology Group Phase I Consortium Study (ADVL1112). Clin. Cancer Res. 2013, 19, 6578–6584. [Google Scholar] [CrossRef]
- Chiappori, A.A.; Kolevska, T.; Spigel, D.R.; Hager, S.; Rarick, M.; Gadgeel, S.; Blais, N.; Von Pawel, J.; Hart, L.; Reck, M.; et al. A Randomized Phase II Study of the Telomerase Inhibitor Imetelstat as Maintenance Therapy for Advanced Non-Small-Cell Lung Cancer. Ann. Oncol. 2015, 26, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Bryan, C.; Rice, C.; Hoffman, H.; Harkisheimer, M.; Sweeney, M.; Skordalakes, E. Structural Basis of Telomerase Inhibition by the Highly Specific BIBR1532. Structure 2015, 23, 1934–1942. [Google Scholar] [CrossRef]
- El-Daly, H.; Kull, M.; Zimmermann, S.; Pantic, M.; Waller, C.F.; Martens, U.M. Selective Cytotoxicity and Telomere Damage in Leukemia Cells Using the Telomerase Inhibitor BIBR1532. Blood 2005, 105, 1742–1749. [Google Scholar] [CrossRef]
- Tauchi, T.; Shin-ya, K.; Sashida, G.; Sumi, M.; Okabe, S.; Ohyashiki, J.H.; Ohyashiki, K. Telomerase Inhibition with a Novel G-Quadruplex-Interactive Agent, Telomestatin: In Vitro and in Vivo Studies in Acute Leukemia. Oncogene 2006, 25, 5719–5725. [Google Scholar] [CrossRef]
- Huppert, J.L.; Balasubramanian, S. Prevalence of Quadruplexes in the Human Genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef]
- Moye, A.L.; Porter, K.C.; Cohen, S.B.; Phan, T.; Zyner, K.G.; Sasaki, N.; Lovrecz, G.O.; Beck, J.L.; Bryan, T.M. Telomeric G-Quadruplexes Are a Substrate and Site of Localization for Human Telomerase. Nat. Commun. 2015, 6, 7643. [Google Scholar] [CrossRef]
- Mender, I.; Gryaznov, S.; Dikmen, Z.G.; Wright, W.E.; Shay, J.W. Induction of Telomere Dysfunction Mediated by the Telomerase Substrate Precursor 6-Thio-2’-Deoxyguanosine. Cancer Discov. 2015, 5, 82–95. [Google Scholar] [CrossRef]
- Zeng, X.; Hernandez-Sanchez, W.; Xu, M.; Whited, T.L.; Baus, D.; Zhang, J.; Berdis, A.J.; Taylor, D.J. Administration of a Nucleoside Analog Promotes Cancer Cell Death in a Telomerase-Dependent Manner. Cell Rep. 2018, 23, 3031–3041. [Google Scholar] [CrossRef]
- Mender, I.; LaRanger, R.; Luitel, K.; Peyton, M.; Girard, L.; Lai, T.-P.; Batten, K.; Cornelius, C.; Dalvi, M.P.; Ramirez, M.; et al. Telomerase-Mediated Strategy for Overcoming Non-Small Cell Lung Cancer Targeted Therapy and Chemotherapy Resistance. Neoplasia 2018, 20, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Sobo, M.; Lee, K.; Senthil Kumar, S.; White, A.R.; Mender, I.; Fuller, C.; Chow, L.M.L.; Fouladi, M.; Shay, J.W.; et al. Induced Telomere Damage to Treat Telomerase Expressing Therapy-Resistant Pediatric Brain Tumors. Mol. Cancer Ther. 2018, 17, 1504–1514. [Google Scholar] [CrossRef]
- Zhang, G.; Wu, L.W.; Mender, I.; Barzily-Rokni, M.; Hammond, M.R.; Ope, O.; Cheng, C.; Vasilopoulos, T.; Randell, S.; Sadek, N.; et al. Induction of Telomere Dysfunction Prolongs Disease Control of Therapy-Resistant Melanoma. Clin. Cancer Res. 2018, 24, 4771–4784. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Uribe, P.; Adrianzen-Ruesta, M.P.; Deng, Z.; Echevarria-Vargas, I.; Mender, I.; Saheb, S.; Liu, Q.; Altieri, D.C.; Murphy, M.E.; Shay, J.W.; et al. Exploiting TERT Dependency as a Therapeutic Strategy for NRAS-Mutant Melanoma. Oncogene 2018, 37, 4058–4072. [Google Scholar] [CrossRef]
- Tagliamonte, M.; Petrizzo, A.; Napolitano, M.; Luciano, A.; Arra, C.; Maiolino, P.; Izzo, F.; Tornesello, M.L.; Aurisicchio, L.; Ciliberto, G.; et al. Novel Metronomic Chemotherapy and Cancer Vaccine Combinatorial Strategy for Hepatocellular Carcinoma in a Mouse Model. Cancer Immunol. Immunother. 2015, 64, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Masutomi, K.; Kyo, S.; Hashimoto, M.; Maida, Y.; Kanaya, T.; Tanaka, M.; Hahn, W.C.; Inoue, M. Efficient Inhibition of Human Telomerase Reverse Transcriptase Expression by RNA Interference Sensitizes Cancer Cells to Ionizing Radiation and Chemotherapy. Hum. Gene Ther. 2005, 16, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Romaniuk-Drapała, A.; Totoń, E.; Konieczna, N.; Machnik, M.; Barczak, W.; Kowal, D.; Kopczyński, P.; Kaczmarek, M.; Rubiś, B. HTERT Downregulation Attenuates Resistance to DOX, Impairs FAK-Mediated Adhesion, and Leads to Autophagy Induction in Breast Cancer Cells. Cells 2021, 10, 867. [Google Scholar] [CrossRef] [PubMed]
- Longe, H.O.; Romesser, P.B.; Rankin, A.M.; Faller, D.V.; Eller, M.S.; Gilchrest, B.A.; Denis, G.V. Telomere Homolog Oligonucleotides Induce Apoptosis in Malignant but Not in Normal Lymphoid Cells: Mechanism and Therapeutic Potential. Int. J. Cancer 2009, 124, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Cheng, J.; Pang, Q.; Wei, X.; Zhang, X.; Wang, P.; Yuan, Z.; Qian, D. BIBR1532, a Selective Telomerase Inhibitor, Enhances Radiosensitivity of Non-Small Cell Lung Cancer Through Increasing Telomere Dysfunction and ATM/CHK1 Inhibition. Int. J. Radiat. Oncol. Biol. Phys. 2019, 105, 861–874. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| No | Type of TERT Treatment | Combination Therapy | Potential Benefits | Reference |
|---|---|---|---|---|
| 1 | hTERT DNA vaccines | Immune checkpoint blockade (CTLA-4 and PD-1) | Immune checkpoint inhibitor potentially modifies the immune regulatory environment of tumors (including expansion of T cells) to synergize with TERT DNA vaccines, leading to synergistic anti-cancer activity | Duperret et al., 2018 [148] |
| 2 | hTERT epitope peptide vaccine | Metronomic chemotherapy | Combinatorial approach increases specific T cell response and decreased Treg frequency | Tagliamonte et al., 2015 [165] |
| 3 | Oncolytic adenovirus (Telomelysin) | Immune checkpoint blockade (anti-PD-1/anti-PD-L1) | Prior treatment with telomelysin sensitizes cancer cells to subsequent immune checkpoint inhibitors administration, which synergises increased tumor regression | Kanaya et al., 2018 [151] |
| 4 | Retroviral-delivered hTERT-specific siRNA | Chemotherapy (topoisomerase inhibitors or bleomycin) and ionizing radiation (IR) | siRNA treatment induced replicative senescence, decreased telomerase activity, attenuated tumorigenicity, and sensitized cancer cells to chemotherapeutic agents and IR | Nakamura et al., 2005 [166] |
| 5 | Lentiviral-delivered hTERT-specific shRNA | Doxorubicin (topo isomerase 2 inhibitor) | hTERT shRNA sensitized cancer cells to doxorubicin through decreased proliferation rates and autophagy | Romaniuk-Drapała et al., 2021 [167] |
| 6 | Nucleoside analogue (6-thio-dG) | Mitochondrial Hsp90 inhibitor (Gamitrinib) | Nucleoside analogue treatment with 6-thio-dG triggers a mitochondrial antioxidant adaptive response, which can be counteracted with mitochondrial Hsp90 inhibitor for higher apoptotic effect in cancer cells | Reyes-Uribe et al., 2018 [164] |
| 7 | Telomere Homolog Oligonucleotide (T-Oligos) | Chemotherapy (Vincristine, Cyclophosphamide, Adriamycin, Prednisone) | Reduced combinational doses of T-oligos and chemotherapy stimulate cell cycle arrest, senescence, and apoptosis via caspase-3 and p53 activation. | Longe et al., 2009 [168] |
| 8 | Small molecule hTERT inhibitor (BIBR1532) | Imatinib mesylate (IM) (tyrosine kinase inhibitor) | IM has been determined as an effective drug against CML but potentiates leukemogenesis via STAT5 phosphorylation leading to TERT activation. Co-administration of TERT inhibitor such as BIBR1532 could ameliorate this side effect. | Celeghin et al., 2016 & Yamada et al., 2011 [91,94] |
| 9 | Small molecule hTERT inhibitor (BIBR1532) | Doxorubicin (topo isomerase 2 inhibitor) | Induction of synergistic anti-cancer effect via p73 activation, repression of proliferation by p21-mediated G1 arrest, downregulation of c-Myc, TERT and survivin, increase ROS leading to caspase-dependent apoptosis, and enhancing pro-oxidant effect of doxorubicin. | Bashash et al., 2017 [141] |
| 10 | Small molecule hTERT inhibitor (BIBR1532) | Ionizing radiation (IR) | BIBR1532 increased radiosensitivity of cancers by enhancing IR-mediated mitotic catastrophe, senescence, and apoptosis via induction of telomere dysfunction and inhibit the ATM/CHK1 pathway | Ding et al., 2019 [169] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yik, M.Y.; Azlan, A.; Rajasegaran, Y.; Rosli, A.; Yusoff, N.M.; Moses, E.J. Mechanism of Human Telomerase Reverse Transcriptase (hTERT) Regulation and Clinical Impacts in Leukemia. Genes 2021, 12, 1188. https://doi.org/10.3390/genes12081188
Yik MY, Azlan A, Rajasegaran Y, Rosli A, Yusoff NM, Moses EJ. Mechanism of Human Telomerase Reverse Transcriptase (hTERT) Regulation and Clinical Impacts in Leukemia. Genes. 2021; 12(8):1188. https://doi.org/10.3390/genes12081188
Chicago/Turabian StyleYik, Mot Yee, Adam Azlan, Yaashini Rajasegaran, Aliaa Rosli, Narazah Mohd Yusoff, and Emmanuel Jairaj Moses. 2021. "Mechanism of Human Telomerase Reverse Transcriptase (hTERT) Regulation and Clinical Impacts in Leukemia" Genes 12, no. 8: 1188. https://doi.org/10.3390/genes12081188
APA StyleYik, M. Y., Azlan, A., Rajasegaran, Y., Rosli, A., Yusoff, N. M., & Moses, E. J. (2021). Mechanism of Human Telomerase Reverse Transcriptase (hTERT) Regulation and Clinical Impacts in Leukemia. Genes, 12(8), 1188. https://doi.org/10.3390/genes12081188