
Hydroxyurea—The Good, the Bad and the Ugly



Abstract
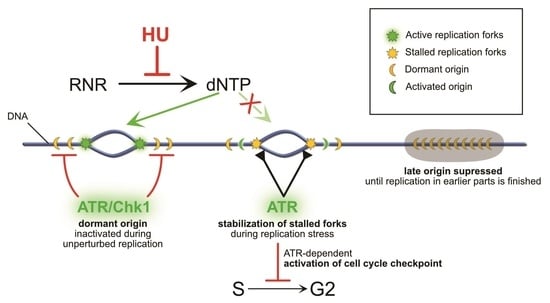
{kind=link}
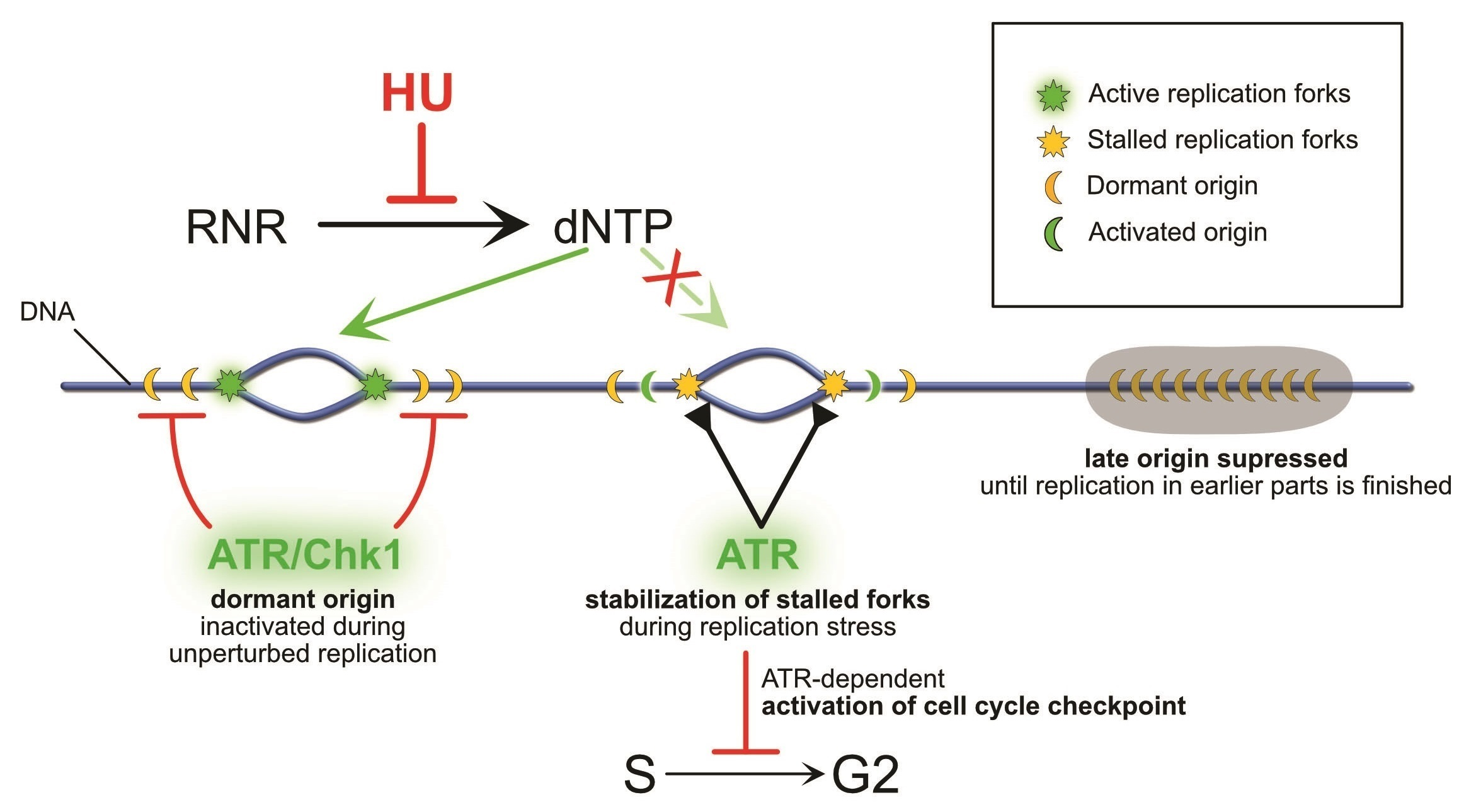
{kind=link}
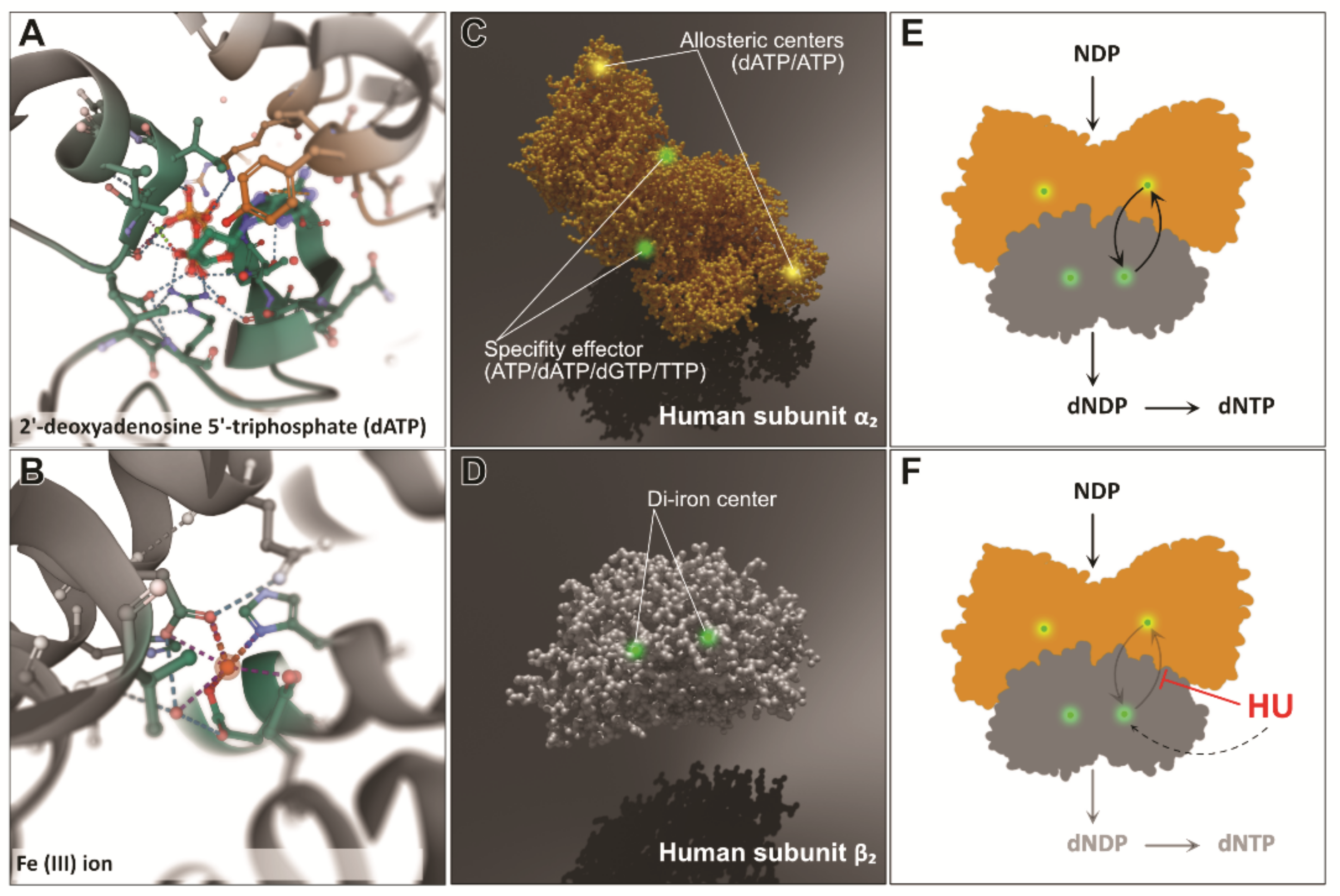
{kind=link}
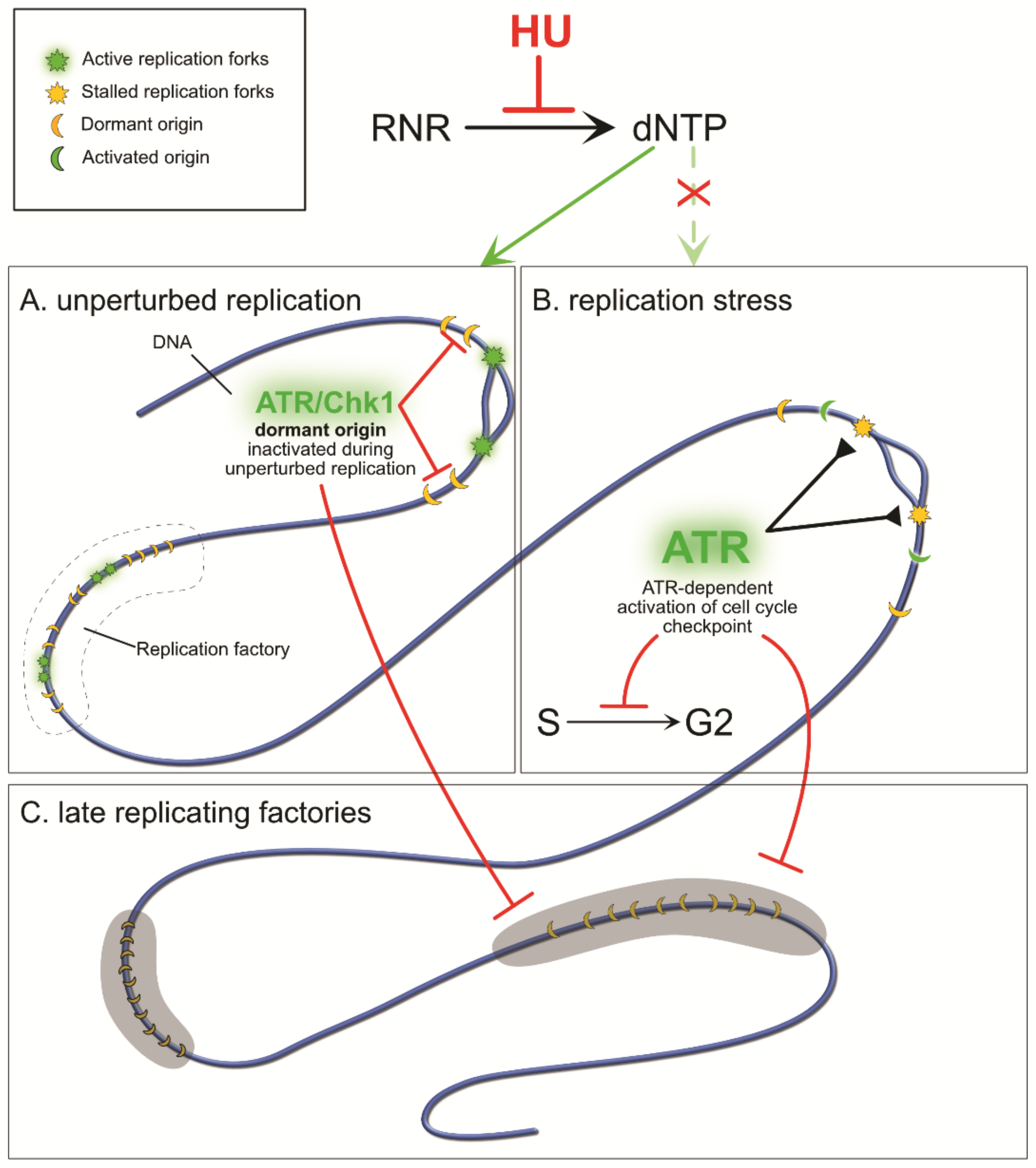
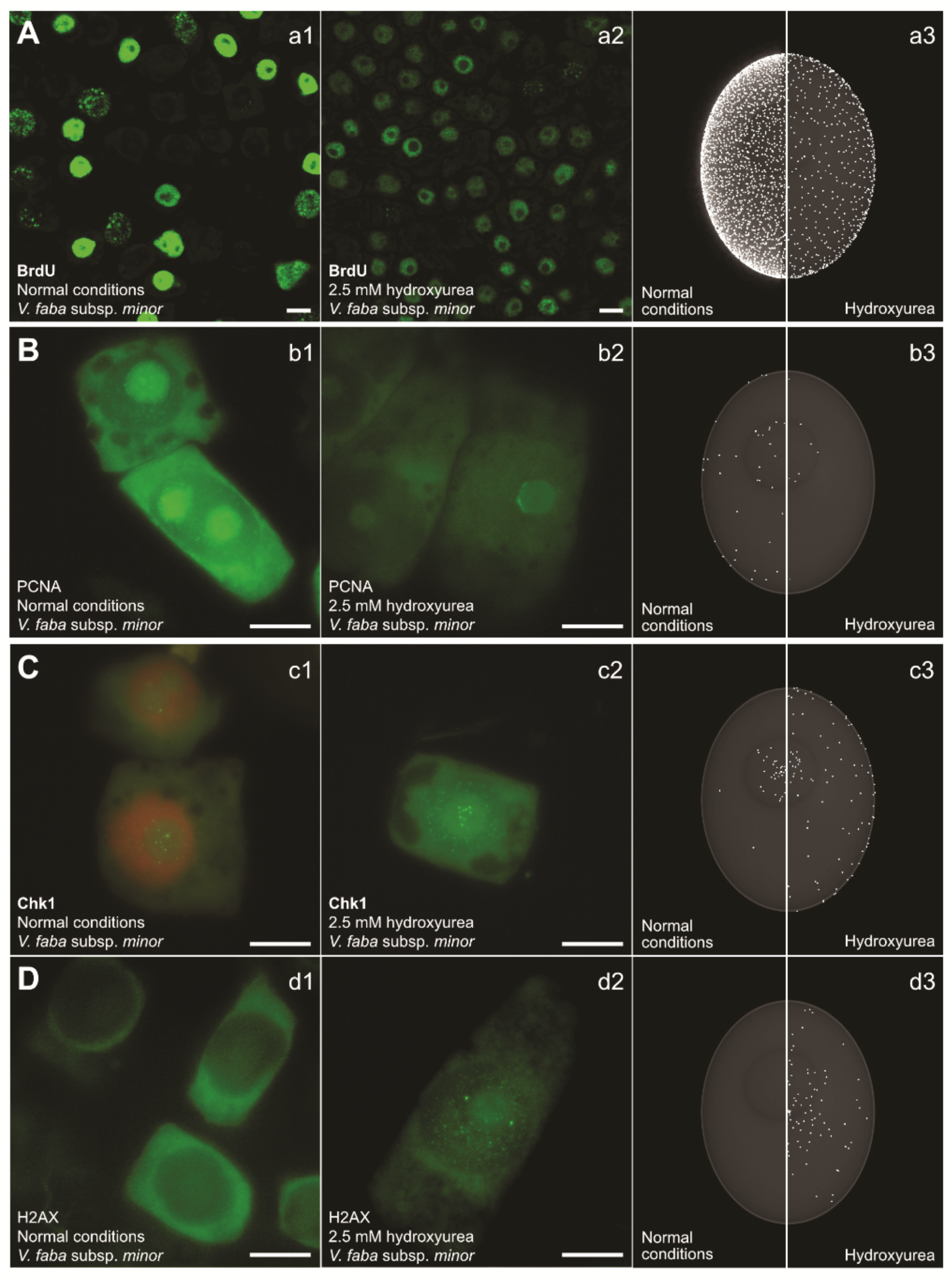{kind=link}
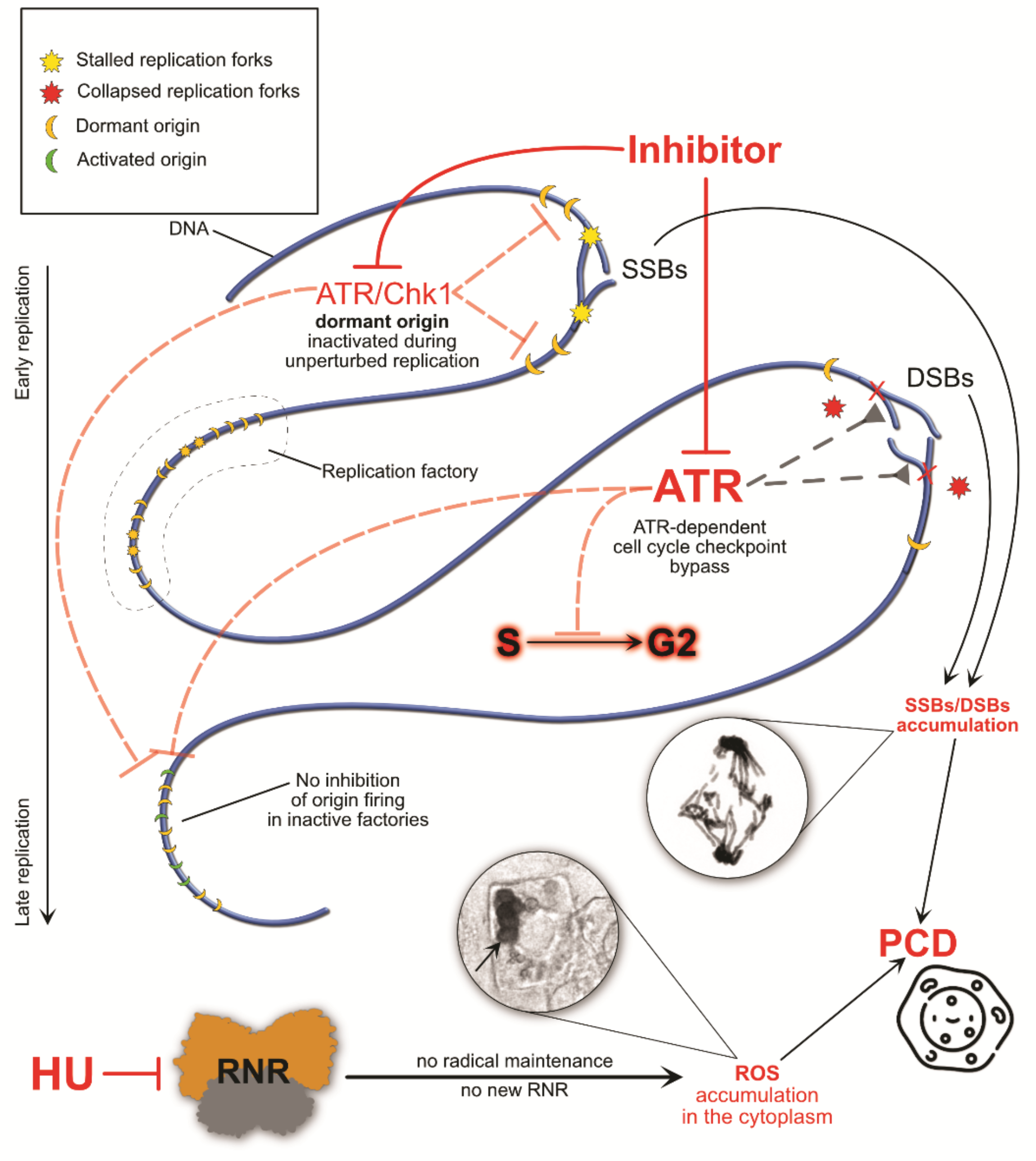{kind=link}
{kind=link}
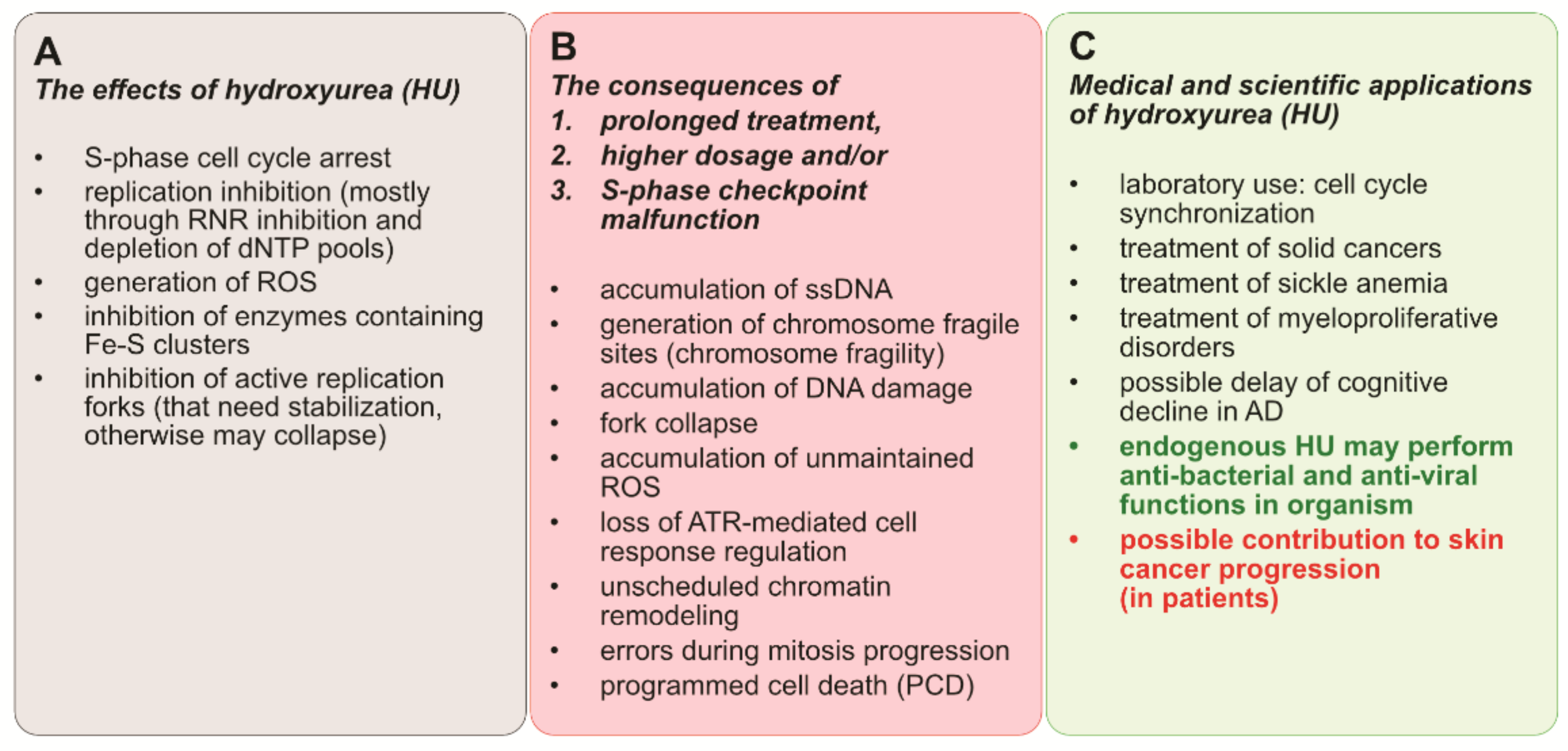{kind=link}
1. Introduction
2. The Mechanism of RNR Inhibition by Hydroxyurea
3. Stalled Fork Maintenance, Cell Cycle Checkpoint Activation, and Production of New dNTPs in Response to HU
4. The Consequences of S-Phase Checkpoint Malfunction
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AO | Acridine orange |
| ATR | Ataxia telangiectasia and Rad3-related |
| BLM | Bloom syndrome protein |
| CF | Caffeine |
| Chk1 | Checkpoint kinase 1 |
| dNTP | Deoxyribonucleoside triphosphate |
| DSB | Double DNA strand break |
| EB | Ethidium bromide |
| Exo1 | Exonuclease 1 |
| HU | Hydroxyurea |
| MCM | Minichromosome maintenance |
| PCET | Proton-coupled electron transfer |
| PCNA | Proliferating cell nuclear antigen |
| RIFT-IR | Reaction-induced FT-IR |
| RNR | Ribonucleotide reductase |
| ROS | Reactive oxygen species |
| RPA | Replication protein A |
| RT | Radical transfer |
References
- Rosenthal, F.; Wislicki, L.; Kollek, L. Über die Beziehungen von Schwersten Blutgiften zu Abbauprodukten des Eiweiss—Ein Beitrag zum Entstehungsmechanismus der perniziösen Anämie. Klin. Wochenschr. 1928, 7, 972–977. [Google Scholar] [CrossRef]
- Adamson, R.H. Activity of Congeners of Hydroxyurea Against Advanced Leukemia L1210. Proc. Soc. Exp. Biol. Med. 1965, 119, 456–458. [Google Scholar] [CrossRef]
- Stearns, B.; Losee, K.A.; Bernstein, J. Hydroxyurea: A New Type of Potential Antitumor Agent. J. Med. Chem. 1963, 6, 201. [Google Scholar] [CrossRef]
- Madaan, K.; Kaushik, D.; Verma, T. Hydroxyurea: A key player in cancer chemotherapy. Expert Rev. Anticancer Ther. 2012, 12, 19–29. [Google Scholar] [CrossRef]
- Spivak, J.L.; Hasselbalch, H. Hydroxycarbamide: A user’s guide for chronic myeloproliferative disorders. Expert Rev. Anticancer Ther. 2011, 11, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Yogev, O.; Anzi, S.; Inoue, K.; Shaulian, E. Induction of transcriptionally active Jun proteins regulates drug-induced senescence. J. Biol. Chem. 2006, 281, 34475–34483. [Google Scholar] [CrossRef] [PubMed]
- Nevitt, S.J.; Jones, A.P.; Howard, J. Hydroxyurea (hydroxycarbamide) for sickle cell disease. Cochrane Database Syst. Rev. 2017, 4, CD002202. [Google Scholar] [CrossRef] [PubMed]
- Tshilolo, L.; Tomlinson, G.; Williams, T.N.; Santos, B.; Olupot-Olupot, P.; Lane, A.; Aygun, B.; Stuber, S.E.; Latham, T.S.; McGann, P.T.; et al. Hydroxyurea for Children with Sickle Cell Anemia in Sub-Saharan Africa. N. Engl. J. Med. 2019, 380, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Brose, R.D.; Lehrmann, E.; Zhang, Y.; Reeves, R.H.; Smith, K.D.; Mattson, M.P. Hydroxyurea attenuates oxidative, metabolic, and excitotoxic stress in rat hippocampal neurons and improves spatial memory in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 72, 121–133. [Google Scholar] [CrossRef]
- Koç, A.; Wheeler, L.J.; Mathews, C.K.; Merrill, G.F. Hydroxyurea Arrests DNA Replication by a Mechanism that Preserves Basal dNTP Pools. J. Biol. Chem. 2004, 279, 223–230. [Google Scholar] [CrossRef]
- Berniak, K.; Rybak, P.; Bernas, T.; Zarebski, M.; Biela, E.; Zhao, H.; Darzynkiewicz, Z.; Dobrucki, J.W. Relationship between DNA damage response, initiated by camptothecin or oxidative stress, and DNA replication, analyzed by quantitative 3D image analysis. Cytom. Part. A 2013, 83, 913–924. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Busby, E.C.; Tibbetts, R.S.; Roos, P.; Taya, Y.; Karnitz, L.M.; Abraham, R.T. Inhibition of ATM and ATR Kinase Activities by the Radiosensitizing Agent, Caffeine. Cancer Res. 1999, 59, 4375–4382. [Google Scholar]
- Boddy, M.N.; Russell, P. DNA replication checkpoint. Curr. Biol. 2001, 11, R953–R956. [Google Scholar] [CrossRef]
- Rybaczek, D. Ultrastructural changes associated with the induction of premature chromosome condensation in Vicia faba root meristem cells. Plant. Cell Rep. 2014, 33, 1547–1564. [Google Scholar] [CrossRef]
- Xu, Y.J.; Singh, A.; Alter, G.M. Hydroxyurea induces cytokinesis arrest in cells expressing a mutated sterol-14α-demethylase in the ergosterol biosynthesis pathway. Genetics 2016, 204, 959–973. [Google Scholar] [CrossRef]
- Huang, M.E.; Facca, C.; Fatmi, Z.; Baïlle, D.; Bénakli, S.; Vernis, L. DNA replication inhibitor hydroxyurea alters Fe-S centers by producing reactive oxygen species in vivo. Sci. Rep. 2016, 6, 29361. [Google Scholar] [CrossRef] [PubMed]
- King, S.B. Nitric oxide production from hydroxyurea. Free Radic. Biol. Med. 2004, 37, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Trovesi, C.; Manfrini, N.; Falcettoni, M.; Longhese, M.P. Regulation of the DNA damage response by cyclin-dependent kinases. J. Mol. Biol. 2013, 425, 4756–4766. [Google Scholar] [CrossRef]
- Huh, M.S.; Ivanochko, D.; Hashem, L.E.; Curtin, M.; Delorme, M.; Goodall, E.; Yan, K.; Picketts, D.J. Stalled replication forks within heterochromatin require ATRX for protection. Cell Death Dis. 2016, 7, e2220-12. [Google Scholar] [CrossRef] [PubMed]
- Giannattasio, M.; Branzei, D. S-phase checkpoint regulations that preserve replication and chromosome integrity upon dNTP depletion. Cell. Mol. Life Sci. 2017, 74, 2361–2380. [Google Scholar] [CrossRef]
- Moiseeva, T.N.; Yin, Y.; Calderon, M.J.; Qian, C.; Schamus-Haynes, S.; Sugitani, N.; Osmanbeyoglu, H.U.; Rothenberg, E.; Watkins, S.C.; Bakkenist, C.J. An ATR and CHK1 kinase signaling mechanism that limits origin firing during unperturbed DNA replication. Proc. Natl. Acad. Sci. USA 2019, 116, 13374–13383. [Google Scholar] [CrossRef]
- Nair, J.; Huang, T.T.; Murai, J.; Haynes, B.; Steeg, P.S.; Pommier, Y.; Lee, J.M. Resistance to the CHK1 inhibitor prexasertib involves functionally distinct CHK1 activities in BRCA wild-type ovarian cancer. Oncogene 2020, 39, 5520–5535. [Google Scholar] [CrossRef] [PubMed]
- Julius, J.; Peng, J.; McCulley, A.; Caridi, C.; Arnak, R.; See, C.; Nugent, C.I.; Feng, W.; Bachant, J. Inhibition of spindle extension through the yeast S phase checkpoint is coupled to replication fork stability and the integrity of centromeric DNA. Mol. Biol. Cell 2019, 30, 2771–2789. [Google Scholar] [CrossRef]
- Shao, J.; Zhou, B.; Chu, B.; Yen, Y. Ribonucleotide Reductase Inhibitors and Future Drug Design. Curr. Cancer Drug Targets 2006, 6, 409–431. [Google Scholar] [CrossRef] [PubMed]
- Sethy, S.; Panda, T.; Jena, R.K. Beneficial Effect of Low Fixed Dose of Hydroxyurea in Vaso-occlusive Crisis and Transfusion Requirements in Adult HbSS Patients: A Prospective Study in a Tertiary Care Center. Indian J. Hematol. Blood Transfus. 2018, 34, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Yazinski, S.A.; Zou, L. Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway. Annu. Rev. Genet. 2016, 50, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Ashley, A.K.; Shrivastav, M.; Nie, J.; Amerin, C.; Troksa, K.; Glanzer, J.G.; Liu, S.; Opiyo, S.O.; Dimitrova, D.D.; Le, P.; et al. DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair 2014, 21, 131–139. [Google Scholar] [CrossRef]
- Kramara, J.; Osia, B.; Malkova, A. Break Induced Replication: The where, the why, and the how. HHS Public Access. Trends Genet. 2018, 34, 518–531. [Google Scholar] [CrossRef]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef]
- Przybyszewski, W.M.; Kasperczyk, J. Rodnikowy mechanizm ubocznej toksyczności hydroksymocznika. Postepy Hig. Med. Dosw. 2006, 60, 516–526. [Google Scholar]
- De Oliveira, E.A.M.; Boy, K.d.A.; Santos, A.P.P.; Machado, C.d.S.; Velloso-Rodrigues, C.; Gerheim, P.S.A.S.; Mendonça, L.M. Evaluation of hydroxyurea genotoxicity in patients with sickle cell disease. Einstein 2019, 17, eAO4742. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, M.; Albergante, L.; Moreno, A.; Carrington, J.T.; Blow, J.J.; Newman, T.J. Inevitability and containment of replication errors for eukaryotic genome lengths spanning megabase to gigabase. Proc. Natl. Acad. Sci. USA 2016, 113, E5765–E5774. [Google Scholar] [CrossRef]
- Elledge, S.J.; Zhou, Z.; Allen, J.B. Ribonucleotide reductase: Regulation, regulation, regulation. Trends Biochem. Sci. 1992, 17, 119–123. [Google Scholar] [CrossRef]
- Offenbacher, A.R.; Barry, B.A. A Proton Wire Mediates Proton Coupled Electron Transfer from Hydroxyurea and Other Hydroxamic Acids to Tyrosyl Radical in Class Ia Ribonucleotide Reductase. J. Phys. Chem. B 2020, 124, 345–354. [Google Scholar] [CrossRef]
- Fairman, J.W.; Wijerathna, S.R.; Ahmad, M.F.; Xu, H.; Nakano, R.; Jha, S.; Prendergast, J.; Welin, R.M.; Flodin, S.; Roos, A.; et al. Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization. Nat. Struct. Mol. Biol. 2011, 18, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Brignole, E.J.; Tsai, K.L.; Chittuluru, J.; Li, H.; Aye, Y.; Penczek, P.A.; Stubbe, J.A.; Drennan, C.L.; Asturias, F. 3.3-Å resolution cryo-EM structure of human ribonucleotide reductase with substrate and allosteric regulators bound. eLife 2018, 7, e31502. [Google Scholar] [CrossRef]
- Eriksson, M.; Uhlin, U.; Ramaswamy, S.; Ekberg, M.; Regnström, K.; Sjöberg, B.M.; Eklund, H. Binding of allosteric effectors to ribonucleotide reductase protein R1: Reduction of active-site cysteines promotes substrate binding. Structure 1997, 5, 1077–1092. [Google Scholar] [CrossRef]
- Rofougaran, R.; Vodnala, M.; Hofer, A. Enzymatically active mammalian ribonucleotide reductase exists primarily as an α6β2 octamer. J. Biol. Chem. 2006, 281, 27705–27711. [Google Scholar] [CrossRef]
- Radivoyevitch, T. Automated mass action model space generation and analysis methods for two-reactant combinatorially complex equilibriums: An analysis of ATP-induced ribonucleotide reductase R1 hexamerization data. Biol. Direct 2009, 4, 50. [Google Scholar] [CrossRef]
- Denysenkov, V.P.; Biglino, D.; Lubitz, W.; Prisner, T.F.; Bennati, M. Structure of the tyrosyl biradical in mouse R2 ribonucleotide reductase from high-field PELDOR. Angew. Chem. Int. Ed. 2008, 47, 1224–1227. [Google Scholar] [CrossRef]
- Minnihan, E.C.; Nocera, D.G.; Stubbe, J. Reversible, long-range radical transfer in E. coli class Ia ribonucleotide reductase. Acc. Chem. Res. 2013, 46, 2524–2535. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Carranza, M.; Jonna, V.R.; Lundin, D.; Sahlin, M.; Carlson, L.A.; Jemal, N.; Högbom, M.; Sjöberg, B.M.; Stenmark, P.; Hofer, A. A ribonucleotide reductase from clostridium botulinum reveals distinct evolutionary pathways to regulation via the overall activity site. J. Biol. Chem. 2020, 295, 15576–15587. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.; Taguchi, A.T.; Stubbe, J.A.; Drennan, C.L. Structure of a trapped radical transfer pathway within a ribonucleotide reductase holocomplex. Science 2020, 368, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Nordlund, P.; Reichard, P. Ribonucleotide reductases. Annu. Rev. Biochem. 2006, 75, 681–706. [Google Scholar] [CrossRef]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velenkar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol*Viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef]
- Rofougaran, R.; Crona, M.; Vodnala, M.; Sjöberg, B.-M.; Hofer, A. Oligomerization status directs overall activity regulation of the Escherichia coli class Ia ribonucleotide reductase. J. Biol. Chem. 2008, 283, 35310–35318. [Google Scholar] [CrossRef]
- Hofer, A.; Crona, M.; Logan, D.T.; Sjöberg, B.-M. DNA building blocks: Keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 50–63. [Google Scholar] [CrossRef]
- Håkansson, P.; Hofer, A.; Thelander, L. Regulation of mammalian ribonucleotide reduction and dNTP pools after DNA damage and in resting cells. J. Biol. Chem. 2006, 281, 7834–7841. [Google Scholar] [CrossRef]
- Sanvisens, N.; De Llanos, R.; Puig, S. Function and regulation of yeast ribonucleotide reductase: Cell cycle, genotoxic stress, and iron bioavailability. Biomed. J. 2013, 36, 51–58. [Google Scholar]
- Guarino, E.; Salguero, I.; Kearsey, S.E. Cellular regulation of ribonucleotide reductase in eukaryotes. Semin. Cell Dev. Biol. 2014, 30, 97–103. [Google Scholar] [CrossRef]
- Lassmann, G.; Thelander, L.; Gräslund, A. EPR stopped-flow studies of the reaction of the tyrosyl radical of protein R2 from ribonucleotide reductase with hydroxyurea. Biochem. Biophys. Res. Commun. 1992, 188, 879–887. [Google Scholar] [CrossRef]
- Singh, A.; Xu, Y.J. The cell killing mechanisms of hydroxyurea. Genes 2016, 7, 99. [Google Scholar] [CrossRef] [PubMed]
- Fontecave, M.; Lepoivre, M.; Elleingand, E.; Gerez, C.; Guittet, O. Resveratrol, a remarkable inhibitor of ribonucleotide reductase. FEBS Lett. 1998, 421, 277–279. [Google Scholar] [CrossRef]
- Rawson, J.M.O.; Roth, M.E.; Xie, J.; Daly, M.B.; Clouser, C.L.; Landman, S.R.; Reilly, C.S.; Bonnac, L.; Kim, B.; Patterson, S.E.; et al. Synergistic reduction of HIV-1 infectivity by 5-azacytidine and inhibitors of ribonucleotide reductase. Bioorganic Med. Chem. 2016, 24, 2410–2422. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, Q.Q.; Lam, C.W.K.; Guo, J.R.; Zhang, W.J.; Wang, C.Y.; Wong, V.K.W.; Yao, M.C.; Zhang, W. Investigation into perturbed nucleoside metabolism and cell cycle for elucidating the cytotoxicity effect of resveratrol on human lung adenocarcinoma epithelial cells. Chin. J. Nat. Med. 2019, 17, 608–615. [Google Scholar] [CrossRef]
- Neuhard, J. Studies on the acid-soluble nucleotide pool in Escherichia coli. IV. Effects of hydroxyurea. BBA Sect. Nucleic Acids Protein Synth. 1967, 145, 1–6. [Google Scholar] [CrossRef]
- Morganroth, P.A.; Hanawalt, P.C. Role of DNA replication and repair in thymineless death in Escherichia coli. J. Bacteriol. 2006, 188, 5286–5288. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kuong, K.J.; Kuzminov, A. Cyanide, Peroxide and Nitric Oxide Formation in Solutions of Hydroxyurea Causes Cellular Toxicity and May Contribute to Its Therapeutic Potency. J. Mol. Biol. 2009, 390, 845–862. [Google Scholar] [CrossRef]
- Fraser, D.I.; Liu, K.T.; Reid, B.J.; Hawkins, E.; Sevier, A.; Pyle, M.; Robinson, J.W.; Ouellette, P.H.R.; Ballantyne, J.S. Widespread natural occurrence of hydroxyurea in animals. PLoS ONE 2015, 10, e0142890. [Google Scholar] [CrossRef]
- Hallmark, L.; Almeida, L.E.F.; Kamimura, S.; Smith, M.; Quezado, Z.M.N. Nitric oxide and sickle cell disease—Is there a painful connection? Exp. Biol. Med. 2021, 246, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Grant, G.D.; Cook, J.G. The Temporal Regulation of S Phase Proteins during G1. Adv. Exp. Med. Biol. 2017, 1042, 335–369. [Google Scholar] [CrossRef]
- Kaykov, A.; Nurse, P. The spatial and temporal organization of origin firing during the S-phase of fission yeast. Genome Res. 2015, 25, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Shoaib, M.; Walter, D.; Gillespie, P.J.; Izard, F.; Fahrenkrog, B.; Lleres, D.; Lerdrup, M.; Johansen, J.V.; Hansen, K.; Julien, E.; et al. Histone H4K20 methylation mediated chromatin compaction threshold ensures genome integrity by limiting DNA replication licensing. Nat. Commun. 2018, 9, 3704. [Google Scholar] [CrossRef] [PubMed]
- Blow, J.J. Defects in the origin licensing checkpoint stresses cells exiting G0. J. Cell Biol. 2019, 218, 2080–2081. [Google Scholar] [CrossRef]
- McIntosh, D.; Blow, J.J. Dormant origins, the licensing checkpoint, and the response to replicative stresses. Cold Spring Harb. Perspect. Biol. 2012, 4, a012955. [Google Scholar] [CrossRef]
- Musiałek, M.W.; Rybaczek, D. Behavior of replication origins in Eukaryota—Spatio-temporal dynamics of licensing and firing. Cell Cycle 2015, 14, 2251–2264. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, J.A.; Gravells, P.; Bryant, H.E. DNA Fiber Assay for the Analysis of DNA Replication Progression in Human Pluripotent Stem Cells. Curr. Protoc. Stem Cell Biol. 2020, 54, e115. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Xu, X.; Yuan, Y.; Zhang, B.; Li, Z.; Xie, Y.; Yan, R.; Zheng, Z.; Ji, J.; et al. The intra-S phase checkpoint directly regulates replication elongation to preserve the integrity of stalled replisomes. Proc. Natl. Acad. Sci USA 2021, 118, e2019183118. [Google Scholar] [CrossRef]
- Nitani, N.; Nakamura, K.; Nakagawa, C.; Masukata, H.; Nakagawa, T. Regulation of DNA replication machinery by Mrc1 in fission yeast. Genetics 2006, 174, 155–165. [Google Scholar] [CrossRef][Green Version]
- Mukherjee, C.; Tripathi, V.; Manolika, E.M.; Heijink, A.M.; Ricci, G.; Merzouk, S.; de Boer, H.R.; Demmers, J.; van Vugt, M.A.T.M.; Ray Chaudhuri, A. RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat. Commun. 2019, 10, 3287. [Google Scholar] [CrossRef]
- Jasencakova, Z.; Scharf, A.N.D.; Ask, K.; Corpet, A.; Imhof, A.; Almouzni, G.; Groth, A. Replication Stress Interferes with Histone Recycling and Predeposition Marking of New Histones. Mol. Cell 2010, 37, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Nazaretyan, S.A.; Savic, N.; Sadek, M.; Hackert, B.J.; Courcelle, J.; Courcelle, C.T. Replication rapidly recovers and continues in the presence of hydroxyurea in Escherichia coli. J. Bacteriol. 2018, 200, e00713-17. [Google Scholar] [CrossRef]
- Ivessa, A.S.; Lenzmeier, B.A.; Bessler, J.B.; Goudsouzian, L.K.; Schnakenberg, S.L.; Zakian, V.A. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol. Cell 2003, 12, 1525–1536. [Google Scholar] [CrossRef]
- Ercilla, A.; Feu, S.; Aranda, S.; Llopis, A.; Brynjólfsdóttir, S.H.; Sørensen, C.S.; Toledo, L.I.; Agell, N. Acute hydroxyurea-induced replication blockade results in replisome components disengagement from nascent DNA without causing fork collapse. Cell. Mol. Life Sci. 2019, 77, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Gardner, N.J.; Gillespie, P.J.; Carrington, J.T.; Shanks, E.J.; McElroy, S.P.; Haagensen, E.J.; Frearson, J.A.; Woodland, A.; Blow, J.J. The High-Affinity Interaction between ORC and DNA that Is Required for Replication Licensing Is Inhibited by 2-Arylquinolin-4-Amines. Cell Chem. Biol. 2017, 24, 981–992.e4. [Google Scholar] [CrossRef] [PubMed]
- Heidinger-Pauli, J.M.; Ünal, E.; Guacci, V.; Koshland, D. The Kleisin Subunit of Cohesin Dictates Damage-Induced Cohesion. Mol. Cell 2008, 31, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar] [CrossRef]
- Menolfi, D.; Delamarre, A.; Lengronne, A.; Pasero, P.; Branzei, D. Essential Roles of the Smc5/6 Complex in Replication through Natural Pausing Sites and Endogenous DNA Damage Tolerance. Mol. Cell 2015, 60, 835–846. [Google Scholar] [CrossRef]
- Weinert, T.A.; Kiser, G.L.; Hartwell, L.H. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev. 1994, 8, 652–665. [Google Scholar] [CrossRef]
- Zuilkoski, C.M.; Skibbens, R.V. PCNA antagonizes cohesin-dependent roles in genomic stability. PLoS ONE 2020, 15, e0235103. [Google Scholar] [CrossRef]
- Van, C.; Yan, S.; Michael, W.M.; Waga, S.; Cimprich, K.A. Continued primer synthesis at stalled replication forks contributes to checkpoint activation. J. Cell Biol. 2010, 189, 233–246. [Google Scholar] [CrossRef]
- Byun, T.S.; Pacek, M.; Yee, M.C.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Iyer, D.R.; Rhind, N. Replication Fork Slowing and Stalling are Distinct, Checkpoint-Independent Consequences of Replicating Damaged DNA. PLoS Genet. 2017, 13, e1006958. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chabes, A.; Domkin, V.; Thelander, L.; Rothstein, R. The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. EMBO J. 2001, 20, 3544–3553. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Muller, E.G.D.; Rothstein, R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol. Cell 1998, 2, 329–340. [Google Scholar] [CrossRef]
- Zhang, Z.; Reese, J.C. Molecular Genetic Analysis of the Yeast Repressor Rfx1/Crt1 Reveals a Novel Two-Step Regulatory Mechanism. Mol. Cell. Biol. 2005, 25, 7399–7411. [Google Scholar] [CrossRef] [PubMed]
- Woolstencroft, R.N.; Bellharz, T.H.; Cook, M.A.; Preiss, T.; Durocher, D.; Tyers, M. Ccr4 contributes to tolerance of replication stress through control of CRT1 mRNA poly(A) tail length. J. Cell Sci. 2006, 119, 5178–5192. [Google Scholar] [CrossRef]
- Buisson, R.; Boisvert, J.L.; Benes, C.H.; Zou, L. Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol. Cell 2015, 59, 1011–1024. [Google Scholar] [CrossRef]
- Koppenhafer, S.L.; Goss, K.L.; Terry, W.W.; Gordon, D.J. Inhibition of the ATR-CHK1 pathway in ewing sarcoma cells causes DNA damage and apoptosis via the CDK2-mediated degradation of RRM2. Mol. Cancer Res. 2020, 18, 91–104. [Google Scholar] [CrossRef]
- Lopez-Contreras, A.J.; Specks, J.; Barlow, J.H.; Ambrogio, C.; Desler, C.; Vikingsson, S.; Rodrigo-Perez, S.; Green, H.; Rasmussen, L.J.; Murga, M.; et al. Increased Rrm2 gene dosage reduces fragile site breakage and prolongs survival of ATR mutant mice. Genes Dev. 2015, 29, 690–695. [Google Scholar] [CrossRef]
- Zou, L.; Elledge, S.J. Sensing DNA Damage Through ATRIP Recognition of RPA-ssDNA Complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Frattini, C.; Villa-Hernández, S.; Pellicanò, G.; Jossen, R.; Katou, Y.; Shirahige, K.; Bermejo, R. Cohesin Ubiquitylation and Mobilization Facilitate Stalled Replication Fork Dynamics. Mol. Cell 2017, 68, 758–772.e4. [Google Scholar] [CrossRef] [PubMed]
- Litwin, I.; Pilarczyk, E.; Wysocki, R. The emerging role of cohesin in the DNA damage response. Genes 2018, 9, 581. [Google Scholar] [CrossRef]
- Rhodes, J.D.P.; Haarhuis, J.H.I.; Grimm, J.B.; Rowland, B.D.; Lavis, L.D.; Nasmyth, K.A. Cohesin Can Remain Associated with Chromosomes during DNA Replication. Cell Rep. 2017, 20, 2749–2755. [Google Scholar] [CrossRef] [PubMed]
- Bot, C.; Pfeiffer, A.; Giordano, F.; Manjeera, D.E.; Dantuma, N.P.; Ström, L. Independent mechanisms recruit the cohesin loader protein NIPBL to sites of DNA damage. J. Cell Sci. 2017, 130, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. XATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088. [Google Scholar] [CrossRef]
- Hoffman, E.A.; McCulley, A.; Haarer, B.; Arnak, R.; Feng, W. Break-seq reveals hydroxyurea-induced chromosome fragility as a result of unscheduled conflict between DNA replication and transcription. Genome Res. 2015, 25, 402–412. [Google Scholar] [CrossRef]
- Żabka, A.; Polit, J.T.; Maszewski, J. DNA replication stress induces deregulation of the cell cycle events in root meristems of Allium cepa. Ann. Bot. 2012, 110, 1581–1591. [Google Scholar] [CrossRef] [PubMed]
- Rybaczek, D.; Musialek, M.W.; Balcerczyk, A. Caffeine-induced premature chromosome condensation results in the apoptosis-like programmed cell death in root meristems of Vicia faba. PLoS ONE 2015, 10, e0142307. [Google Scholar] [CrossRef] [PubMed]
- Rybaczek, D.; Musiałek, M.W.; Vrána, J.; Petrovská, B.; Pikus, E.G.; Doležel, J. Kinetics of DNA Repair in Vicia faba Meristem Regeneration Following Replication Stress. Cells 2021, 10, 88. [Google Scholar] [CrossRef]
- Palumbo, E.; Matricardi, L.; Tosoni, E.; Bensimon, A.; Russo, A. Replication dynamics at common fragile site FRA6E. Chromosoma 2010, 119, 575–587. [Google Scholar] [CrossRef]
- Hashash, N.; Johnson, A.L.; Cha, R.S. Regulation of fragile sites expression in budding yeast by MEC1, RRM3 and hydroxyurea. J. Cell Sci. 2011, 124, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Técher, H.; Koundrioukoff, S.; Carignon, S.; Wilhelm, T.; Millot, G.A.; Lopez, B.S.; Brison, O.; Debatisse, M. Signaling from Mus81-Eme2-Dependent DNA Damage Elicited by Chk1 Deficiency Modulates Replication Fork Speed and Origin Usage. Cell Rep. 2016, 14, 1114–1127. [Google Scholar] [CrossRef]
- Cortez, D. Replication-Coupled DNA Repair. Mol. Cell 2019, 74, 866–876. [Google Scholar] [CrossRef]
- Shiu, J.L.; Wu, C.K.; Chang, S.B.; Sun, Y.J.; Chen, Y.J.; Lai, C.C.; Chiu, W.T.; Chang, W.T.; Myung, K.; Su, W.P.; et al. The HLTF–PARP1 interaction in the progression and stability of damaged replication forks caused by methyl methanesulfonate. Oncogenesis 2020, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.B.; Cortez, D. Fork reversal, too much of a good thing. Cell Cycle 2014, 13, 1049–1050. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.H.; Rothstein, R. Rad52 recruitment is DNA replication independent and regulated by Cdc28 and the Mec1 kinase. EMBO J. 2009, 28, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Fumasoni, M.; Zwicky, K.; Vanoli, F.; Lopes, M.; Branzei, D. Error-Free DNA Damage Tolerance and Sister Chromatid Proximity during DNA Replication Rely on the Polα/Primase/Ctf4 Complex. Mol. Cell 2015, 57, 812–823. [Google Scholar] [CrossRef]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Mutreja, K.; Krietsch, J.; Hess, J.; Ursich, S.; Berti, M.; Roessler, F.K.; Zellweger, R.; Patra, M.; Gasser, G.; Lopes, M. ATR-Mediated Global Fork Slowing and Reversal Assist Fork Traverse and Prevent Chromosomal Breakage at DNA Interstrand Cross-Links. Cell Rep. 2018, 24, 2629–2642.e5. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.E.; Ajazi, A.; Carotenuto, W.; Foiani, M.; Giannattasio, M. Rad53-Mediated Regulation of Rrm3 and Pif1 DNA Helicases Contributes to Prevention of Aberrant Fork Transitions under Replication Stress. Cell Rep. 2015, 13, 80–92. [Google Scholar] [CrossRef]
- Couch, F.B.; Bansbach, C.E.; Driscoll, R.; Luzwick, J.W.; Glick, G.G.; Bétous, R.; Carroll, C.M.; Jung, S.Y.; Qin, J.; Cimprich, K.A.; et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013, 27, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Ragland, R.L.; Patel, S.; Rivard, R.S.; Smith, K.; Peters, A.A.; Bielinsky, A.K.; Brown, E.J. RNF4 and PLK1 are required for replication fork collapse in ATR-deficient cells. Genes Dev. 2013, 27, 2259–2273. [Google Scholar] [CrossRef]
- Yu, C.; Gan, H.; Han, J.; Zhou, Z.X.; Jia, S.; Chabes, A.; Farrugia, G.; Ordog, T.; Zhang, Z. Strand-Specific Analysis Shows Protein Binding at Replication Forks and PCNA Unloading from Lagging Strands when Forks Stall. Mol. Cell 2014, 56, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Katou, Y.; Nakato, R.; Shirahige, K.; Donaldson, A.D. Replication-Coupled PCNA Unloading by the Elg1 Complex Occurs Genome-wide and Requires Okazaki Fragment Ligation. Cell Rep. 2015, 12, 774–787. [Google Scholar] [CrossRef]
- Ohashi, E.; Tsurimoto, T. Functions of multiple clamp and clamp-loader complexes in eukaryotic DNA replication. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2017; Volume 1042, pp. 135–162. [Google Scholar]
- Errico, A.; Aze, A.; Costanzo, V. Mta2 promotes Tipin-dependent maintenance of replication fork integrity. Cell Cycle 2014, 13, 2120–2128. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.; Neelsen, K.J.; Lukas, J. Molecular Cell Perspective Replication Catastrophe: When a Checkpoint Fails because of Exhaustion. Mol. Cell. 2017, 66, 735–749. [Google Scholar] [CrossRef]
- Segurado, M.; Diffley, J.F.X. Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev. 2008, 22, 1816–1827. [Google Scholar] [CrossRef]
- Kaochar, S.; Shanks, L.; Weinert, T. Checkpoint genes and Exo1 regulate nearby inverted repeat fusions that form dicentric chromosomes in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2010, 107, 21605–21610. [Google Scholar] [CrossRef]
- Morafraile, E.C.; Bugallo, A.; Carreira, R.; Fernández, M.; Martín-Castellanos, C.; Blanco, M.G.; Segurado, M. Exo1 phosphorylation inhibits exonuclease activity and prevents fork collapse in rad53 mutants independently of the 14-3-3 proteins. Nucleic Acids Res. 2020, 48, 3053–3070. [Google Scholar] [CrossRef]
- Tomimatsu, N.; Mukherjee, B.; Harris, J.L.; Boffo, F.L.; Hardebeck, M.C.; Potts, P.R.; Khanna, K.K.; Burma, S. DNA-damage-induced degradation of EXO1 exonuclease limits DNA end resection to ensure accurate DNA repair. J. Biol. Chem. 2017, 292, 10779–10790. [Google Scholar] [CrossRef]
- Hae, Y.Y.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Mcm2 is a direct substrate of ATM and ATR during DNA damage and DNA replication checkpoint responses. J. Biol. Chem. 2004, 279, 53353–53364. [Google Scholar] [CrossRef]
- Shorrocks, A.M.K.; Jones, S.E.; Tsukada, K.; Morrow, C.A.; Belblidia, Z.; Shen, J.; Vendrell, I.; Fischer, R.; Kessler, B.M.; Blackford, A.N. The Bloom syndrome complex senses RPA-coated single-stranded DNA to restart stalled replication forks. Nat. Commun. 2021, 12, 585. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.L.; North, P.S.; Hickson, I.D. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat. Struct. Mol. Biol. 2007, 14, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Franchitto, A.; Pichierri, P. Bloom’s syndrome protein is required for correct relocalization of RAD50/MRE11/NBS1 complex after replication fork arrest. J. Cell Biol. 2002, 157, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Godoy, V.G.; Jarosz, D.F.; Walker, F.L.; Simmons, L.A.; Walker, G.C. Y-family DNA polymerases respond to DNA damage-independent inhibition of replication fork progression. EMBO J. 2006, 25, 868–879. [Google Scholar] [CrossRef]
- Imlay, J.A.; Chin, S.M.; Linn, S. Toxic DNA damage by hydrogen peroxide through the fenton reaction in vivo and in vitro. Science 1988, 240, 640–642. [Google Scholar] [CrossRef]
- Rowe, L.A.; Degtyareva, N.; Doetsch, P.W. DNA damage-induced reactive oxygen species (ROS) stress response in Saccharomyces cerevisiae. Free Radic. Biol. Med. 2008, 45, 1167–1177. [Google Scholar] [CrossRef]
- Rowe, L.A.; Degtyareva, N.; Doetsch, P.W. Yap1: A DNA damage responder in Saccharomyces cerevisiae. Mech. Ageing Dev. 2012, 133, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Jiang, W.; Krebs, C.; Stubbe, J. YfaE, a ferredoxin involved in diferric-tyrosyl radical maintenance in Escherichia coli ribonucleotide reductase. Biochemistry 2007, 46, 11577–11588. [Google Scholar] [CrossRef]
- Nakayashiki, T.; Mori, H. Genome-Wide screening with hydroxyurea reveals a link between nonessential ribosomal proteins and reactive oxygen species production. J. Bacteriol. 2013, 195, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Netz, D.J.A.; Stith, C.M.; Stümpfih, M.; Köpf, G.; Vogel, D.; Genau, H.M.; Stodola, J.L.; Lill, R.; Burgers, P.M.J.; Pierik, A.J. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat. Chem. Biol. 2011, 8, 125–132. [Google Scholar] [CrossRef]
- Rudolf, J.; Makrantoni, V.; Ingledew, W.J.; Stark, M.J.R.; White, M.F. The DNA repair helicases XPD and FancJ have essential iron-sulfur domains. Mol. Cell 2006, 26, 801–808. [Google Scholar] [CrossRef]
- Holt, M.E.; Salay, L.E.; Chazin, W.J. A Polymerase With Potential: The Fe–S Cluster in Human DNA Primase. Methods Enzymol. 2017, 595, 361–390. [Google Scholar]
- Liu, L.; Huang, M. Essential role of the iron-sulfur cluster binding domain of the primase regulatory subunit Pri2 in DNA replication initiation. Protein Cell 2015, 6, 194–210. [Google Scholar] [CrossRef]
- Kettani, T.; Cotton, F.; Gulbis, B.; Ferster, A.; Kumps, A. Plasma hydroxyurea determined by gas chromatography-mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 446–450. [Google Scholar] [CrossRef]
- Scott, D.K.; Neville, K.; Garg, U. Determination of hydroxyurea in serum or plasma using gas chromatography-mass spectrometry (GC-MS). Methods Mol. Biol. 2010, 603, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Adragna, N.C.; Fonseca, P.; Lauf, P.K. Hydroxyurea affects cell morphology, cation transport, and red blood cell adhesion in cultured vascular endothelial cells. Blood 1994, 83, 553–560. [Google Scholar] [CrossRef]
- Cokic, V.P.; Smith, R.D.; Beleslin-Cokic, B.B.; Njoroge, J.M.; Miller, J.L.; Gladwin, M.T.; Schechter, A.N. Hydroxyurea induces fetal hemoglobin by the nitric oxide–dependent activation of soluble guanylyl cyclase. J. Clin. Investig. 2003, 111, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Cantisani, C.; Kiss, N.; Naqeshbandi, A.F.; Tosti, G.; Tofani, S.; Cartoni, C.; Carmosino, I.; Cantoresi, F. Nonmelanoma skin cancer associated with Hydroxyurea treatment: Overview of the literature and our own experience. Dermatol. Ther. 2019, 32, e13043. [Google Scholar] [CrossRef] [PubMed]
- Contreras Castillo, S.; Montibus, B.; Rocha, A.; Duke, W.; von Meyenn, F.; McLornan, D.; Harrison, C.; Mullally, A.; Schulz, R.; Oakey, R.J. Hydroxycarbamide effect on DNA methylation and gene expression in myeloproliferative neoplasms. Genome Res. 2021, 9, gr-27006. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musiałek, M.W.; Rybaczek, D. Hydroxyurea—The Good, the Bad and the Ugly. Genes 2021, 12, 1096. https://doi.org/10.3390/genes12071096
Musiałek MW, Rybaczek D. Hydroxyurea—The Good, the Bad and the Ugly. Genes. 2021; 12(7):1096. https://doi.org/10.3390/genes12071096
Chicago/Turabian StyleMusiałek, Marcelina W., and Dorota Rybaczek. 2021. "Hydroxyurea—The Good, the Bad and the Ugly" Genes 12, no. 7: 1096. https://doi.org/10.3390/genes12071096
APA StyleMusiałek, M. W., & Rybaczek, D. (2021). Hydroxyurea—The Good, the Bad and the Ugly. Genes, 12(7), 1096. https://doi.org/10.3390/genes12071096