
Alternative RNA Splicing—The Trojan Horse of Cancer Cells in Chemotherapy



 , and
, and
Abstract
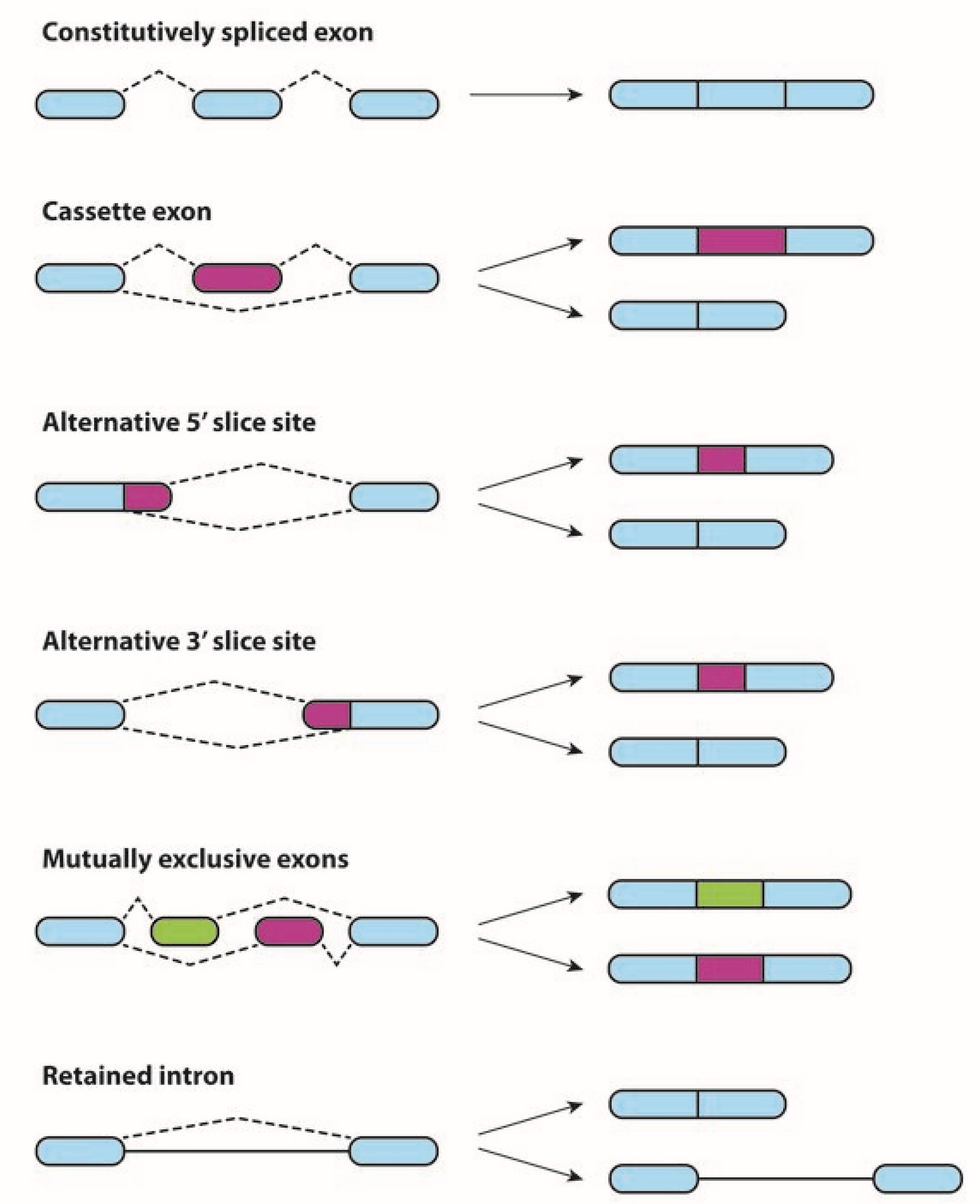
1. Alternative Splicing in Physiological and Neoplastic Processes
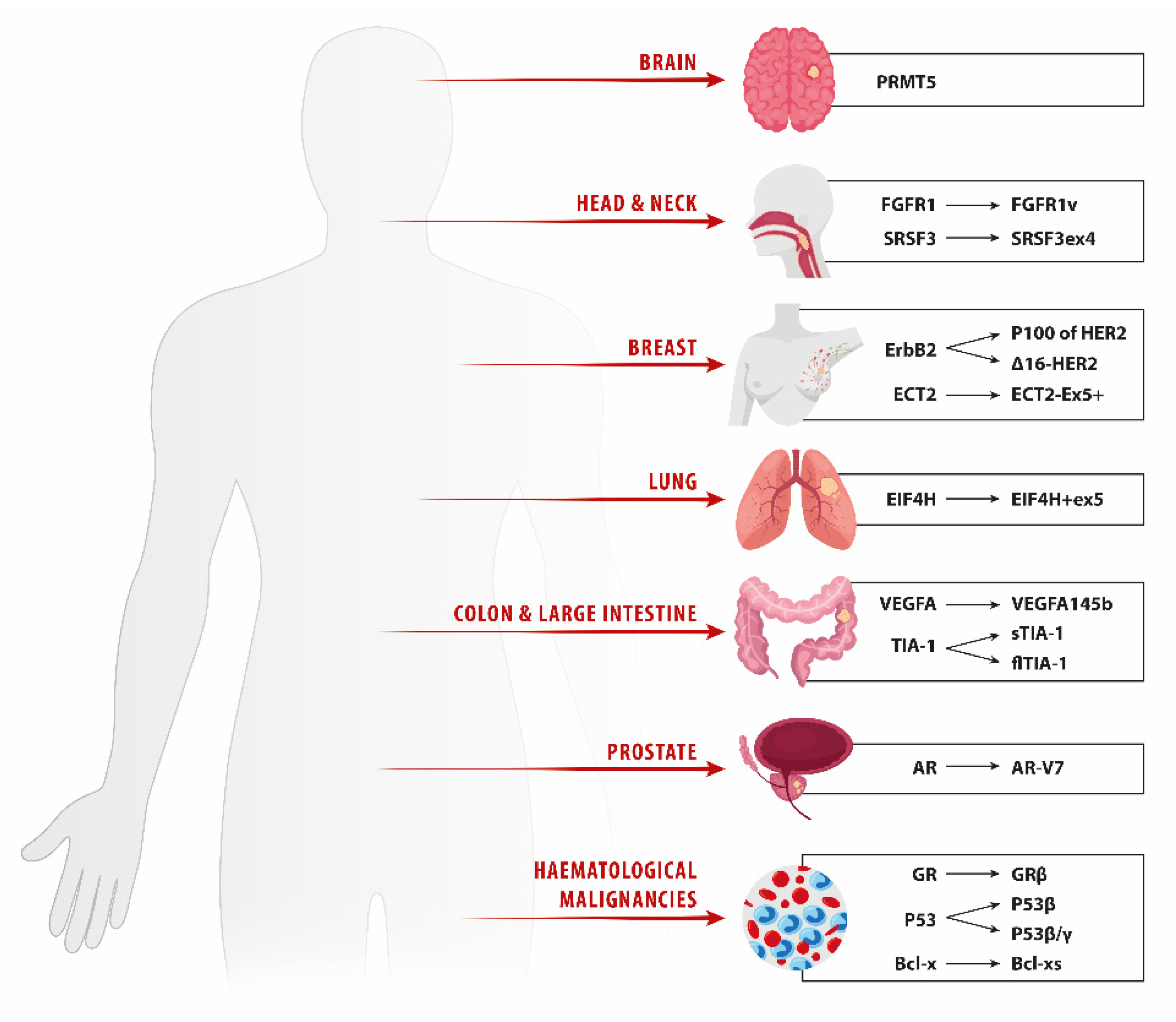
2. Lung Cancer
Anticancer Drug Resistance Based on AS in Lung Cancer
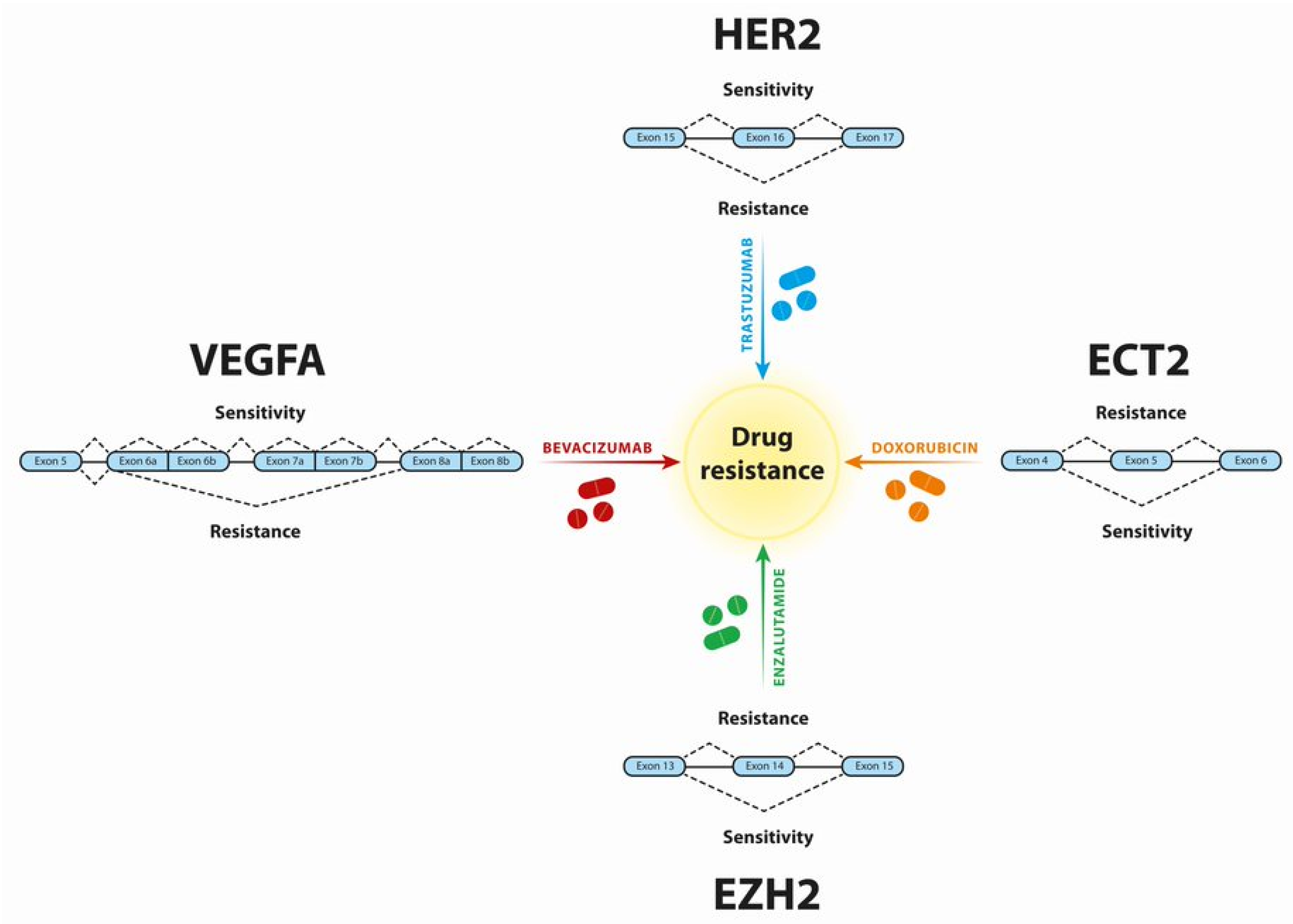
3. Breast Cancer
4. Prostate Cancer
5. Head and Neck Squamous Cell Carcinoma
6. Glioma
7. Metastatic Colorectal Carcinoma
8. Hematologic Malignancies
8.1. AS in Lymphoid Malignancies
8.2. AS in Myeloid Malignancies
9. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shampo, M.A.; Kyle, R.A. Phillip Sharp—Nobel Prize for Discovery of “Split Genes”. Mayo Clin. Proc. 2004, 79, 727. [Google Scholar] [CrossRef]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.; Kopylov, A.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The Size of the Human Proteome: The Width and Depth. Int. J. Anal. Chem. 2016, 2016, 7436849. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nat. Cell Biol. 2010, 463, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Early, P.; Rogers, J.; Davis, M.; Calame, K.; Bond, M.; Wall, R.; Hood, L. Two mRNAs can be produced from a single immunoglobulin μ gene by alternative RNA processing pathways. Cell 1980, 20, 313–319. [Google Scholar] [CrossRef]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef]
- Pohl, M.; Bortfeldt, R.H.; Grützmann, K.; Schuster, S. Alternative splicing of mutually exclusive exons—A review. Biosystems 2013, 114, 31–38. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Huang, B.O.; Xu, Y.-M.; Huang, L.-F.; Lin, J.; Zhang, J.; Min, Q.-H.; Yang, W.-M.; Wang, X.-Z. Mechanism of alternative splicing and its regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nat. Cell Biol. 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Bowler, E.; Oltean, S. Alternative Splicing in Angiogenesis. Int. J. Mol. Sci. 2019, 20, 2067. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Lynch, K.W. Regulation of Alternative Splicing by Signal Transduction Pathways. Adv. Exp. Med. Biol. 2007, 623, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Black, D.L. A CaMK IV responsive RNA element mediates depolarization-induced alternative splicing of ion channels. Nature 2001, 410, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Li, Y.; Yang, X.; Wu, Y.-P.; Lin, L.-J.; Liu, X.-M. Significance of alternative splicing in cancer cells. Chin. Med. J. 2020, 133, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qian, J.; Gu, C.; Yang, Y. Alternative splicing and cancer: A systematic review. Signal Transduct. Target. Ther. 2021, 6, 78. [Google Scholar] [CrossRef]
- Belluti, S.; Rigillo, G.; Imbriano, C. Transcription Factors in Cancer: When Alternative Splicing Determines Opposite Cell Fates. Cells 2020, 9, 760. [Google Scholar] [CrossRef]
- Bonnal, S.C.; López-Oreja, I.; Valcárcel, J. Roles and mechanisms of alternative splicing in cancer-implications for care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef]
- Oka, M.; Xu, L.; Suzuki, T.; Yoshikawa, T.; Sakamoto, H.; Uemura, H.; Yoshizawa, A.C.; Suzuki, Y.; Nakatsura, T.; Ishihama, Y.; et al. Aberrant splicing isoforms detected by full-length transcriptome sequencing as transcripts of potential neoantigens in non-small cell lung cancer. Genome Biol. 2021, 22, 9. [Google Scholar] [CrossRef] [PubMed]
- Cherry, S.; Lynch, K.W. Alternative splicing and cancer: Insights, opportunities, and challenges from an expanding view of the transcriptome. Genes Dev. 2020, 34, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Shin, J.-H.; Stuelten, C.H.; Zhang, Y.E. TGF-β-induced alternative splicing of TAK1 promotes EMT and drug resistance. Oncogene 2019, 38, 3185–3200. [Google Scholar] [CrossRef]
- Brown, R.L.; Reinke, L.M.; Damerow, M.S.; Perez, D.; Chodosh, L.A.; Yang, J.; Cheng, C. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J. Clin. Investig. 2011, 121, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.J.; Bates, D. VEGF-A splicing: The key to anti-angiogenic therapeutics? Nat. Rev. Cancer 2008, 8, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Carpenter, R.; Han, W.; Lo, H.-W. The GLI1 splice variant TGLI1 promotes glioblastoma angiogenesis and growth. Cancer Lett. 2014, 343, 51–61. [Google Scholar] [CrossRef]
- Jurica, M.S.; Mesecar, A.; Heath, P.J.; Shi, W.; Nowak, T.; Stoddard, B.L. The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure 1998, 6, 195–210. [Google Scholar] [CrossRef]
- Han, J.; Li, J.; Ho, J.C.; Chia, G.S.; Kato, H.; Jha, S.; Yang, H.; Poellinger, L.; Lee, K.L. Hypoxia is a Key Driver of Alternative Splicing in Human Breast Cancer Cells. Sci. Rep. 2017, 7, 4108. [Google Scholar] [CrossRef] [PubMed]
- Black, A.; Gamarra, J.; Giudice, J. More than a messenger: Alternative splicing as a therapeutic target. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 194395. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Yan, J.-Q.; Liu, M.; Ma, Y.-L.; Le, K.-D.; Dong, B.; Li, G.-H. Development of alternative splicing signature in lung squamous cell carcinoma. Med. Oncol. 2021, 38, 49. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Lee, K.-W.; Srivastava, R.M.; Kuo, F.; Krishna, C.; Chowell, D.; Makarov, V.; Hoen, D.; Dalin, M.G.; Wexler, L.; et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019, 25, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Mano, H. Non-solid oncogenes in solid tumors:EML4-ALKfusion genes in lung cancer. Cancer Sci. 2008, 99, 2349–2355. [Google Scholar] [CrossRef] [PubMed]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Zhang, J.-Z.; Zhao, R.-L.; Yang, L.; Guo, D. PSMD7 downregulation induces apoptosis and suppresses tumorigenesis of esophageal squamous cell carcinoma via the mTOR/p70S6K pathway. FEBS Open Bio 2018, 8, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, N.; Lu, Z.; Sun, S.; Huang, J.; Chen, Z.; He, J. Prognostic alternative mRNA splicing signature in non-small cell lung cancer. Cancer Lett. 2017, 393, 40–51. [Google Scholar] [CrossRef]
- Zhang, S.; Bao, Y.; Shen, X.; Pan, Y.; Sun, Y.; Xiao, M.; Chen, K.; Wei, H.; Zuo, J.; Saffen, D.; et al. RNA binding motif protein 10 suppresses lung cancer progression by controlling alternative splicing of eukaryotic translation initiation factor 4H. EBioMedicine 2020, 61, 103067. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cidlowski, J. Cellular Processing of the Glucocorticoid Receptor Gene and Protein: New Mechanisms for Generating Tissue-specific Actions of Glucocorticoids. J. Biol. Chem. 2011, 286, 3177–3184. [Google Scholar] [CrossRef]
- Sciarrillo, R.; Wojtuszkiewicz, A.; Kooi, I.E.; Leon, L.G.; Sonneveld, E.; Kuiper, R.P.; Jansen, G.; Giovannetti, E.; Kaspers, G.J.; Cloos, J. Glucocorticoid Resistant Pediatric Acute Lymphoblastic Leukemia Samples Display Altered Splicing Profile and Vulnerability to Spliceosome Modulation. Cancers 2020, 12, 723. [Google Scholar] [CrossRef]
- Adesso, L.; Calabretta, S.; Barbagallo, F.; Capurso, G.; Pilozzi, E.; Geremia, R.; Fave, G.D.; Sette, C. Gemcitabine triggers a pro-survival response in pancreatic cancer cells through activation of the MNK2/eIF4E pathway. Oncogene 2012, 32, 2848–2857. [Google Scholar] [CrossRef]
- Buxade, M.; Parra-Palau, J.L.; Proud, C.G. The Mnks: MAP kinase-interacting kinases (MAP kinase signal-integrating kinases). Front. Biosci. 2008, 13, 5359–5373. [Google Scholar] [CrossRef]
- Scheper, G.C.; Parra, J.L.; Wilson, M.; van Kollenburg, B.; Vertegaal, A.; Han, Z.-G.; Proud, C.G. The N and C Termini of the Splice Variants of the Human Mitogen-Activated Protein Kinase-Interacting Kinase Mnk2 Determine Activity and Localization. Mol. Cell. Biol. 2003, 23, 5692–5705. [Google Scholar] [CrossRef] [PubMed]
- Calabretta, S.; Bielli, P.; Passacantilli, I.; Pilozzi, E.; Fendrich, V.; Capurso, G.; Fave, G.D.; Sette, C. Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells. Oncogene 2016, 35, 2031–2039. [Google Scholar] [CrossRef]
- Aird, D.; Teng, T.; Huang, C.-L.; Pazolli, E.; Banka, D.; Cheung-Ong, K.; Eifert, C.; Furman, C.; Wu, Z.J.; Seiler, M.; et al. Sensitivity to splicing modulation of BCL2 family genes defines cancer therapeutic strategies for splicing modulators. Nat. Commun. 2019, 10, 137. [Google Scholar] [CrossRef]
- Tauro, B.J.; Mathias, R.; Greening, D.; Gopal, S.K.; Ji, H.; Kapp, E.A.; Coleman, B.M.; Hill, A.; Kusebauch, U.; Hallows, J.L.; et al. Oncogenic H-Ras Reprograms Madin-Darby Canine Kidney (MDCK) Cell-derived Exosomal Proteins Following Epithelial-Mesenchymal Transition. Mol. Cell. Proteom. 2013, 12, 2148–2159. [Google Scholar] [CrossRef] [PubMed]
- Finci, L.I.; Zhang, X.; Huang, X.; Zhou, Q.; Tsai, J.; Teng, T.; Agrawal, A.; Chan, B.; Irwin, S.; Karr, C.; et al. The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes Dev. 2018, 32, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Zhao, Q.; Zhao, J.; Zhang, W.; Sun, Y.; Qin, P.; Lv, Y.; Bai, L.; Yang, Q.; Chen, L.; et al. SRSF1 modulates PTPMT1 alternative splicing to regulate lung cancer cell radioresistance. EBioMedicine 2018, 38, 113–126. [Google Scholar] [CrossRef]
- Liu, J.; Bhadra, M.; Sinnakannu, J.R.; Yue, W.L.; Tan, C.W.; Rigo, F.; Ong, S.T.; Roca, X. Overcoming imatinib resistance conferred by the BIM deletion polymorphism in chronic myeloid leukemia with splice-switching antisense oligonucleotides. Oncotarget 2017, 8, 77567–77585. [Google Scholar] [CrossRef]
- Cope, C.L.; Gilley, R.; Balmanno, K.; Sale, M.J.; Howarth, K.D.; Hampson, M.; Smith, P.D.; Guichard, S.M.; Cook, S. Adaptation to mTOR kinase inhibitors by amplification of eIF4E to maintain cap-dependent translation. J. Cell Sci. 2014, 127, 788–800. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Francies, F.Z.; Hull, R.; Khanyile, R.; Dlamini, Z. Breast cancer in low-middle income countries: Abnormality in splicing and lack of targeted treatment options. Am. J. Cancer Res. 2020, 10, 1568–1591. [Google Scholar]
- Siegfried, Z.; Karni, R. The role of alternative splicing in cancer drug resistance. Curr. Opin. Genet. Dev. 2018, 48, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Read, A.; Natrajan, R. Splicing dysregulation as a driver of breast cancer. Endocr. Relat. Cancer 2018, 25, R467–R478. [Google Scholar] [CrossRef]
- Buoso, E.; Ronfani, M.; Galasso, M.; Ventura, D.; Corsini, E.; Racchi, M. Cortisol-induced SRSF3 expression promotes GR splicing, RACK1 expression and breast cancer cells migration. Pharmacol. Res. 2019, 143, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Castagnoli, L.; Ladomery, M.; Tagliabue, E.; Pupa, S.M. The d16HER2 Splice Variant: A Friend or Foe of HER2-Positive Cancers? Cancers 2019, 11, 902. [Google Scholar] [CrossRef] [PubMed]
- Volpi, C.C.; Pietrantonio, F.; Gloghini, A.; Fucà, G.; Giordano, S.; Corso, S.; Pruneri, G.; Antista, M.; Cremolini, C.; Fasano, E.; et al. The landscape of d16HER2 splice variant expression across HER2-positive cancers. Sci. Rep. 2019, 9, 3545. [Google Scholar] [CrossRef] [PubMed]
- Hart, V.; Gautrey, H.; Kirby, J.; Tyson-Capper, A. HER2 splice variants in breast cancer: Investigating their impact on diagnosis and treatment outcomes. Oncotarget 2020, 11, 4338–4357. [Google Scholar] [CrossRef]
- Sun, Y.; Yan, L.; Guo, J.; Shao, J.; Jia, R. Downregulation of SRSF3 by antisense oligonucleotides sensitizes oral squamous cell carcinoma and breast cancer cells to paclitaxel treatment. Cancer Chemother. Pharmacol. 2019, 84, 1133–1143. [Google Scholar] [CrossRef]
- Safikhani, Z.; Smirnov, P.; Thu, K.L.; Silvester, J.; El-Hachem, N.; Quevedo, R.; Lupien, M.; Mak, T.W.; Cescon, D.; Haibe-Kains, B. Gene isoforms as expression-based biomarkers predictive of drug response in vitro. Nat. Commun. 2017, 8, 1126. [Google Scholar] [CrossRef]
- Tanaka, I.; Chakraborty, A.; Saulnier, O.; Benoit-Pilven, C.; Vacher, S.; Labiod, D.; Lam, E.W.F.; Bièche, I.; Delattre, O.; Pouzoulet, F.; et al. ZRANB2 and SYF2-mediated splicing programs converging on ECT2 are involved in breast cancer cell resistance to doxorubicin. Nucleic Acids Res. 2020, 48, 2676–2693. [Google Scholar] [CrossRef]
- Wang, B.-D.; Lee, N.H. Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef] [PubMed]
- Iwai, K.; Yaguchi, M.; Nishimura, K.; Yamamoto, Y.; Tamura, T.; Nakata, D.; Dairiki, R.; Kawakita, Y.; Mizojiri, R.; Ito, Y.; et al. Anti-tumor efficacy of a novel CLK inhibitor via targeting RNA splicing and MYC-dependent vulnerability. EMBO Mol. Med. 2018, 10, e8289. [Google Scholar] [CrossRef]
- Huang, G.; Song, C.; Wang, N.; Qin, T.; Sui, S.; Obr, A.; Zeng, L.; Wood, T.L.; Leroith, D.; Li, M.; et al. RNA-binding protein CUGBP1 controls the differential INSR splicing in molecular subtypes of breast cancer cells and affects cell aggressiveness. Carcinogenesis 2020, 41, 1294–1305. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Su, B.; Yu, P.; He, J.; Meng, L.; Xiao, Q.; Sun, J.; Zhou, K.; Xue, Y.; et al. Transcriptome-wide analysis and modelling of prognostic alternative splicing signatures in invasive breast cancer: A prospective clinical study. Sci. Rep. 2020, 10, 16504. [Google Scholar] [CrossRef]
- Gökmen-Polar, Y.; Neelamraju, Y.; Goswami, C.P.; Gu, Y.; Gu, X.; Nallamothu, G.; Vieth, E.; Janga, S.C.; Ryan, M.; Badve, S.S. Splicing factor ESRP 1 controls ER-positive breast cancer by altering metabolic pathways. EMBO Rep. 2019, 20, e46078. [Google Scholar] [CrossRef]
- Pucci, S.; Polidoro, C.; Greggi, C.; Amati, F.; Morini, E.; Murdocca, M.; Biancolella, M.; Orlandi, A.; Sangiuolo, F.; Novelli, G. Pro-oncogenic action of LOX-1 and its splice variant LOX-1Δ4 in breast cancer phenotypes. Cell Death Dis. 2019, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Bowling, E.A.; Wang, J.H.; Gong, F.; Wu, W.; Neill, N.J.; Kim, I.S.; Tyagi, S.; Orellana, M.; Kurley, S.J.; Dominguez-Vidaña, R.; et al. Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer. Cell 2021, 184, 384–403.e21. [Google Scholar] [CrossRef]
- Koedoot, E.; van Steijn, E.; Vermeer, M.; González-Prieto, R.; Vertegaal, A.C.O.; Martens, J.W.M.; Le Dévédec, S.E.; van de Water, B. Splicing factors control triple-negative breast cancer cell mitosis through SUN2 interaction and sororin intron retention. J. Exp. Clin. Cancer Res. 2021, 40, 82. [Google Scholar] [CrossRef]
- Tyson-Capper, A.; Gautrey, H. Regulation of Mcl-1 alternative splicing by hnRNP F, H1 and K in breast cancer cells. RNA Biol. 2018, 15, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhou, Z.; Subhramanyam, C.S.; Cao, Q.; Heng, Z.S.L.; Liu, W.; Fu, X.; Hu, Q. SRPK1 acetylation modulates alternative splicing to regulate cisplatin resistance in breast cancer cells. Commun. Biol. 2020, 3, 268. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhao, S.; Li, X.; Kirk, J.S.; Tang, D.G. Prostate Luminal Progenitor Cells in Development and Cancer. Trends Cancer 2018, 4, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Park, D.; Zhong, Y.; Lu, Y.; Rycaj, K.; Gong, S.; Chen, X.; Liu, X.; Chao, H.-P.; Whitney, P.; et al. Stem cell and neurogenic gene-expression profiles link prostate basal cells to aggressive prostate cancer. Nat. Commun. 2016, 7, 10798. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Hu, Q.; Liu, X.; Ji, Y.; Chao, H.-P.; Liu, Y.; Tracz, A.; Kirk, J.; Buonamici, S.; Zhu, P.; et al. Intron retention is a hallmark and spliceosome represents a therapeutic vulnerability in aggressive prostate cancer. Nat. Commun. 2020, 11, 2089. [Google Scholar] [CrossRef]
- Sveen, A.; Kilpinen, S.; Ruusulehto, A.; Lothe, R.A.; Skotheim, R.I. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35, 2413–2427. [Google Scholar] [CrossRef] [PubMed]
- Yae, T.; Tsuchihashi, K.; Ishimoto, T.; Motohara, T.; Yoshikawa, M.; Yoshida, G.; Wada, T.; Masuko, T.; Mogushi, K.; Tanaka, H.; et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat. Commun. 2012, 3, 883. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.R.; Li, Y.; Xie, N.; Gleave, M.; Cox, M.E.; Collins, C.C.; Dong, X. Alternative RNA splicing of the MEAF6 gene facilitates neuroendocrine prostate cancer progression. Oncotarget 2017, 8, 27966–27975. [Google Scholar] [CrossRef]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.-C.; Wongvipat, J.; Ku, S.-Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef]
- Rajan, P.; Sudbery, I.; Villasevil, M.E.M.; Mui, E.; Fleming, J.; Davis, M.; Ahmad, I.; Edwards, J.; Sansom, O.J.; Sims, D.; et al. Next-generation Sequencing of Advanced Prostate Cancer Treated with Androgen-deprivation Therapy. Eur. Urol. 2014, 66, 32–39. [Google Scholar] [CrossRef]
- Sowalsky, A.; Xia, Z.; Wang, L.; Zhao, H.; Chen, S.; Bubley, G.J.; Balk, S.P.; Li, W. Whole Transcriptome Sequencing Reveals Extensive Unspliced mRNA in Metastatic Castration-Resistant Prostate Cancer. Mol. Cancer Res. 2015, 13, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, A.W.; Mo, F.; Wang, K.; McConeghy, B.; Brahmbhatt, S.; Jong, L.; Mitchell, D.M.; Johnston, R.; Haegert, A.; Li, E.; et al. Heterogeneity in the inter-tumor transcriptome of high risk prostate cancer. Genome Biol. 2014, 15, 426. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Deng, Q.; Chao, H.-P.; Liu, X.; Lu, Y.; Lin, K.; Liu, B.; Tang, G.W.; Zhang, D.; Tracz, A.; et al. Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses. Nat. Commun. 2018, 9, 3600. [Google Scholar] [CrossRef]
- Cao, B.; Qi, Y.; Zhang, G.; Xu, D.; Zhan, Y.; Alvarez, X.; Guo, Z.; Fu, X.; Plymate, S.R.; Sartor, O.; et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget 2014, 5, 1646–1656. [Google Scholar] [CrossRef]
- Munkley, J.; Livermore, K.; Rajan, P.; Elliott, D.J. RNA splicing and splicing regulator changes in prostate cancer pathology. Hum. Genet. 2017, 136, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Seitz, A.K.; Thoene, S.; Bietenbeck, A.; Nawroth, R.; Tauber, R.; Thalgott, M.; Schmid, S.; Secci, R.; Retz, M.; Gschwend, J.E.; et al. AR-V7 in Peripheral Whole Blood of Patients with Castration-resistant Prostate Cancer: Association with Treatment-specific Outcome Under Abiraterone and Enzalutamide. Eur. Urol. 2017, 72, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jia, D.; Kim, H.; Elmageed, Z.Y.A.; Datta, A.; Davis, R.; Srivastav, S.K.; Moroz, K.; Crawford, B.E.; Moparty, K.; et al. Dysregulation of miR-212 Promotes Castration Resistance through hnRNPH1-Mediated Regulation of AR and AR-V7: Implications for Racial Disparity of Prostate Cancer. Clin. Cancer Res. 2016, 22, 1744–1756. [Google Scholar] [CrossRef]
- Nadiminty, N.; Tummala, R.; Liu, C.; Lou, W.; Evans, C.P.; Gao, A.C. NF-κB2/p52:c-Myc:hnRNPA1 Pathway Regulates Expression of Androgen Receptor Splice Variants and Enzalutamide Sensitivity in Prostate Cancer. Mol. Cancer Ther. 2015, 14, 1884–1895. [Google Scholar] [CrossRef]
- Kawamura, N.; Nimura, K.; Saga, K.; Ishibashi, A.; Kitamura, K.; Nagano, H.; Yoshikawa, Y.; Ishida, K.; Nonomura, N.; Arisawa, M.; et al. SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression. Cancer Res. 2019, 79, 5204–5217. [Google Scholar] [CrossRef]
- Will, C.L.; Schneider, C.; Macmillan, A.M.; Katopodis, N.F.; Neubauer, G.; Wilm, M.; Lührmann, R.; Query, C.C. A novel U2 and U11/U12 snRNP protein that associates with the pre-mRNA branch site. EMBO J. 2001, 20, 4536–4546. [Google Scholar] [CrossRef] [PubMed]
- Cretu, C.; Schmitzová, J.; Ponce-Salvatierra, A.; Dybkov, O.; De Laurentiis, E.I.; Sharma, K.; Will, C.L.; Urlaub, H.; Lührmann, R.; Pena, V. Molecular Architecture of SF3b and Structural Consequences of Its Cancer-Related Mutations. Mol. Cell 2016, 64, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Will, C.L.; Urlaub, H.; Achsel, T.; Gentzel, M.; Wilm, M.; Lührmann, R. Characterization of novel SF3b and 17S U2 snRNP proteins, including a human Prp5p homologue and an SF3b DEAD-box protein. EMBO J. 2002, 21, 4978–4988. [Google Scholar] [CrossRef] [PubMed]
- Valcarcel, J.; Gaur, R.K.; Singh, R.; Green, M.R. Interaction of U2AF65 RS Region with Pre-mRNA Branch Point and Promotion of Base Pairing with U2 snRNA. Science 1996, 273, 1706–1709. [Google Scholar] [CrossRef]
- Singh, R.; Valcarcel, J.; Green, M.R. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science 1995, 268, 1173–1176. [Google Scholar] [CrossRef]
- Shao, C.; Yang, B.; Wu, T.; Huang, J.; Tang, P.; Zhou, Y.; Zhou, J.; Qiu, J.; Jiang, L.; Li, H.; et al. Mechanisms for U2AF to define 3′ splice sites and regulate alternative splicing in the human genome. Nat. Struct. Mol. Biol. 2014, 21, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Misra, A.; Ou, J.; Zhu, L.J.; Green, M.R. Global Promotion of Alternative Internal Exon Usage by mRNA 3′ End Formation Factors. Mol. Cell 2015, 58, 819–831. [Google Scholar] [CrossRef]
- Cretu, C.; Agrawal, A.A.; Cook, A.; Will, C.L.; Fekkes, P.; Smith, P.G.; Lührmann, R.; Larsen, N.; Buonamici, S.; Pena, V. Structural Basis of Splicing Modulation by Antitumor Macrolide Compounds. Mol. Cell 2018, 70, 265–273.e8. [Google Scholar] [CrossRef]
- Eskens, F.A.; Ramos, F.J.; Burger, H.; O’Brien, J.P.; Piera, A.; de Jonge, M.J.; Mizui, Y.; Wiemer, E.A.; Carreras, M.J.; Baselga, J.; et al. Phase I Pharmacokinetic and Pharmacodynamic Study of the First-in-Class Spliceosome Inhibitor E7107 in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2013, 19, 6296–6304. [Google Scholar] [CrossRef]
- Ryan, M.; Wong, W.C.; Brown, R.; Akbani, R.; Su, X.; Broom, B.; Melott, J.; Weinstein, J. TCGASpliceSeq a compendium of alternative mRNA splicing in cancer. Nucleic Acids Res. 2016, 44, D1018–D1022. [Google Scholar] [CrossRef]
- Phillips, J.W.; Pan, Y.; Tsai, B.; Xie, Z.; Demirdjian, L.; Xiao, W.; Yang, H.T.; Zhang, Y.; Lin, C.H.; Cheng, D.; et al. Pathway-guided analysis identifies Myc-dependent alternative pre-mRNA splicing in aggressive prostate cancers. Proc. Natl. Acad. Sci. USA 2020, 117, 5269–5279. [Google Scholar] [CrossRef] [PubMed]
- Urbanski, L.M.; Leclair, N.; Anczuków, O. Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip. Rev. RNA 2018, 9, e1476. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Li, C.; McCoy, J.P.; Deng, C.X.; Zheng, Z.M. SRp20 is a proto-oncogene critical for cell proliferation and tumor induction and maintenance. Int. J. Biol. Sci. 2010, 6, 806–826. [Google Scholar] [CrossRef] [PubMed]
- Corbo, C.; Orrù, S.; Salvatore, F. SRp20: An overview of its role in human diseases. Biochem. Biophys. Res. Commun. 2013, 436, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Jumaa, H.; Nielsen, P.J. The splicing factor SRp20 modifies splicing of its own mRNA and ASF/SF2 antagonizes this regulation. EMBO J. 1997, 16, 5077–5085. [Google Scholar] [CrossRef]
- Li, Y.; Donmez, N.; Sahinalp, C.; Xie, N.; Wang, Y.; Xue, H.; Mo, F.; Beltran, H.; Gleave, M.; Wang, Y.; et al. SRRM4 Drives Neuroendocrine Transdifferentiation of Prostate Adenocarcinoma Under Androgen Receptor Pathway Inhibition. Eur. Urol. 2017, 71, 68–78. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, C.; Cui, Y.; Nadiminty, N.; Lou, W.; Gao, A.C. Interleukin-6 induces neuroendocrine differentiation (NED) through suppression of RE-1 silencing transcription factor (REST). Prostate 2014, 74, 1086–1094. [Google Scholar] [CrossRef]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; Macdonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef]
- Zhang, X.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lucas, J.M.; Lam, H.-M.; Dumpit, R.; Corey, E.; Chéry, L.; et al. SRRM4 Expression and the Loss of REST Activity May Promote the Emergence of the Neuroendocrine Phenotype in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2015, 21, 4698–4708. [Google Scholar] [CrossRef]
- Lee, A.R.; Gan, Y.; Tang, Y.; Dong, X. A novel mechanism of SRRM4 in promoting neuroendocrine prostate cancer development via a pluripotency gene network. EBioMedicine 2018, 35, 167–177. [Google Scholar] [CrossRef]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbé, D.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef]
- Yu, J.; Yu, J.; Mani, R.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; et al. An Integrated Network of Androgen Receptor, Polycomb, and TMPRSS2-ERG Gene Fusions in Prostate Cancer Progression. Cancer Cell 2010, 17, 443–454. [Google Scholar] [CrossRef]
- Kim, J.; Lee, Y.; Lu, X.; Song, B.; Fong, K.-W.; Cao, Q.; Licht, J.D.; Zhao, J.C.; Yu, J. Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Rep. 2018, 25, 2808–2820.e4. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhang, Z.; Cheng, L.; Wang, R.; Chen, X.; Kong, Y.; Feng, F.; Ahmad, N.; Li, L.; Liu, X. Inhibition of enhancer of zeste homolog 2 (EZH2) overcomes enzalutamide resistance in castration-resistant prostate cancer. J. Biol. Chem. 2019, 294, 9911–9923. [Google Scholar] [CrossRef]
- Wang, H.-J.; Pochampalli, M.; Wang, L.-Y.; Zou, J.X.; Li, P.-S.; Hsu, S.-C.; Wang, B.-J.; Huang, S.-H.; Yang, P.; Yang, J.C.; et al. KDM8/JMJD5 as a dual coactivator of AR and PKM2 integrates AR/EZH2 network and tumor metabolism in CRPC. Oncogene 2019, 38, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Todorova, K.; Metodiev, M.V.; Metodieva, G.; Mincheff, M.; Fernández, N.; Hayrabedyan, S. Micro-RNA-204 Participates in TMPRSS2/ERG Regulation and Androgen Receptor Reprogramming in Prostate Cancer. Horm. Cancer 2017, 8, 28–48. [Google Scholar] [CrossRef]
- Todorova, K.; Zasheva, D.; Kanev, K.; Hayrabedyan, S. miR-204 Shifts the Epithelial to Mesenchymal Transition in Concert with the Transcription Factors RUNX2, ETS1, and cMYB in Prostate Cancer Cell Line Model. J. Cancer Res. 2014, 2014, 1–14. [Google Scholar] [CrossRef]
- Chen, K.; Xiao, H.; Zeng, J.; Yu, G.; Zhou, H.; Huang, C.; Yao, W.; Xiao, W.; Hu, J.; Guan, W.; et al. Alternative Splicing of EZH2 pre-mRNA by SF3B3 Contributes to the Tumorigenic Potential of Renal Cancer. Clin. Cancer Res. 2017, 23, 3428–3441. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Cohen, E.; LaMonte, S.J.; Erb, N.L.; Beckman, K.L.; Sadeghi, N.; Hutcheson, K.; Stubblefield, M.D.; Abbott, D.M.; Fisher, P.S.; Stein, K.D.; et al. American Cancer Society Head and Neck Cancer Survivorship Care Guideline. CA Cancer J. Clin. 2016, 66, 203–239. [Google Scholar] [CrossRef]
- Rothenberg, S.M.; Ellisen, L.W. The molecular pathogenesis of head and neck squamous cell carcinoma. J. Clin. Investig. 2012, 122, 1951–1957. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.-F.; Liu, B.; Zhang, W.-F.; Zhao, Y.-F.; Kulkarni, A.B.; Sun, Z.-J. Dual induction of apoptotic and autophagic cell death by targeting survivin in head neck squamous cell carcinoma. Cell Death Dis. 2015, 6, e1771. [Google Scholar] [CrossRef]
- Perri, F.; Pacelli, R.; Scarpati, G.D.V.; Cella, L.; Giuliano, M.; Caponigro, F.; Pepe, S. Radioresistance in head and neck squamous cell carcinoma: Biological bases and therapeutic implications. Head Neck 2015, 37, 763–770. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Alshaimaa, A.; Maria, M.V.; Daniel, D.; Herbert, R.; Jozsef, D.; Ira-Ida, S. Epithelial-mesenchymal crosstalk induces radioresistance in HNSCC cells. Oncotarget 2017, 9, 3641–3652. [Google Scholar] [CrossRef]
- You, G.-R.; Cheng, A.-J.; Lee, L.-Y.; Huang, Y.-C.; Liu, H.; Chen, Y.-J.; Chang, J.T. Prognostic signature associated with radioresistance in head and neck cancer via transcriptomic and bioinformatic analyses. BMC Cancer 2019, 19, 64. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Yonekubo, Y.; Hanson, N.; Sastre-Perona, A.; Basin, A.; Rytlewski, J.; Dolgalev, I.; Meehan, S.; Tsirigos, A.; Beronja, S.; et al. TGF-β-Induced Quiescence Mediates Chemoresistance of Tumor-Propagating Cells in Squamous Cell Carcinoma. Cell Stem Cell 2017, 21, 650–664.e8. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Si, S.; Li, X.; Sun, W.; Cui, L. Identification and validation of an alternative splicing-based prognostic signature for head and neck squamous cell carcinoma. J. Cancer 2020, 11, 4571–4580. [Google Scholar] [CrossRef]
- Xing, L.; Zhang, X.; Tong, D. Systematic Profile Analysis of Prognostic Alternative Messenger RNA Splicing Signatures and Splicing Factors in Head and Neck Squamous Cell Carcinoma. DNA Cell Biol. 2019, 38, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Libório, T.N.; Ferreira, E.; Xavier, F.C.A.; Carraro, D.M.; Kowalski, L.P.; Soares, F.A.; Nunes, F.D. TGIF1 splicing variant 8 is overexpressed in oral squamous cell carcinoma and is related to pathologic and clinical behavior. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2013, 116, 614–625. [Google Scholar] [CrossRef]
- Hamid, R.; Patterson, J.; Brandt, S.J. Genomic structure, alternative splicing and expression of TG-interacting factor, in human myeloid leukemia blasts and cell lines. Biochim. Biophys. Acta Bioenerg. 2008, 1779, 347–355. [Google Scholar] [CrossRef]
- Qi, Z.; Wang, F.; Yu, G.; Wang, D.; Yao, Y.; You, M.; Liu, J.; Liu, J.; Sun, Z.; Ji, C.; et al. SRSF1 serves as a critical posttranscriptional regulator at the late stage of thymocyte development. Sci. Adv. 2021, 7, eabf0753. [Google Scholar] [CrossRef]
- Guo, J.; Jia, J.; Jia, R. PTBP1 and PTBP2 impaired autoregulation of SRSF3 in cancer cells. Sci. Rep. 2015, 5, 14548. [Google Scholar] [CrossRef]
- Jia, R.; Zhang, S.; Liu, M.; Zhang, Y.; Liu, Y.; Fan, M.; Guo, J. HnRNP L is important for the expression of oncogene SRSF3 and oncogenic potential of oral squamous cell carcinoma cells. Sci. Rep. 2016, 6, 35976. [Google Scholar] [CrossRef]
- Kurokawa, K.; Akaike, Y.; Masuda, K.; Kuwano, Y.; Nishida, K.; Yamagishi, N.; Kajita, K.; Tanahashi, T.; Rokutan, K. Downregulation of serine/arginine-rich splicing factor 3 induces G1 cell cycle arrest and apoptosis in colon cancer cells. Oncogene 2013, 33, 1407–1417. [Google Scholar] [CrossRef]
- Lin, J.-C.; Lee, Y.-C.; Tan, T.-H.; Liang, Y.-C.; Chuang, H.-C.; Fann, Y.C.; Johnson, K.R.; Lin, Y.-J. RBM4-SRSF3-MAP4K4 splicing cascade modulates the metastatic signature of colorectal cancer cell. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Zhao, L.; Zhang, H.; Feng, X.; Xu, H.; Hao, J.; Wang, S.; Yang, Q.; Zou, L.; Su, X.; et al. Loss of TDP43 inhibits progression of triple-negative breast cancer in coordination with SRSF3. Proc. Natl. Acad. Sci. USA 2018, 115, E3426–E3435. [Google Scholar] [CrossRef]
- Cavaloc, Y.; Bourgeois, C.; Kister, L.; Stévenin, J. The splicing factors 9G8 and SRp20 transactivate splicing through different and specific enhancers. RNA 1999, 5, 468–483. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Allen, M.A.; Larsen, A.; MacMorris, M.; Han, M.; Blumenthal, T. Genes involved in pre-mRNA 3’-end formation and transcription termination revealed by a lin-15 operon Muv suppressor screen. Proc. Natl. Acad. Sci. USA 2008, 105, 16665–16670. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Gattoni, R.; Stévenin, J.; Steitz, J.A. SR Splicing Factors Serve as Adapter Proteins for TAP-Dependent mRNA Export. Mol. Cell 2003, 11, 837–843. [Google Scholar] [CrossRef]
- Do, D.V.; Strauss, B.; Cukuroglu, E.; Macaulay, I.; Wee, K.B.; Hu, T.X.; Igor, R.D.L.M.; Lee, C.; Harrison, A.; Butler, R.; et al. SRSF3 maintains transcriptome integrity in oocytes by regulation of alternative splicing and transposable elements. Cell Discov. 2018, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Nguyen, T.D.; Li, S.; Nguyen, T.A. SRSF3 recruits DROSHA to the basal junction of primary microRNAs. RNA 2018, 24, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.M.; Daijogo, S.; Semler, B.L. A nucleo-cytoplasmic SR protein functions in viral IRES-mediated translation initiation. EMBO J. 2006, 26, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Jumaa, H.; Nielsen, P.J. Regulation of SRp20 exon 4 splicing. Biochim. Biophys. Acta 2000, 1494, 137–143. [Google Scholar] [CrossRef]
- Yang, S.; Jia, R.; Bian, Z. SRSF5 functions as a novel oncogenic splicing factor and is upregulated by oncogene SRSF3 in oral squamous cell carcinoma. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1161–1172. [Google Scholar] [CrossRef]
- Dlamini, Z.; Alaouna, M.; Mbatha, S.; Bhayat, A.; Mabongo, M.; Chatziioannou, A.; Hull, R. Genetic Drivers of Head and Neck Squamous Cell Carcinoma: Aberrant Splicing Events, Mutational Burden, HPV Infection and Future Targets. Genes 2021, 12, 422. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Zhang, J.; Jiang, L.; Wang, Y.; Ren, X.; Cheng, B.; Xia, J. Comprehensive Analysis of Prognostic Alternative Splicing Signatures in Oral Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 1740. [Google Scholar] [CrossRef]
- Papaspyrou, G.; Hoch, S.; Rinaldo, A.; Rodrigo, J.P.; Takes, R.P.; Van Herpen, C.; Werner, J.A.; Ferlito, A.; Tapia, J.P.R. Chemotherapy and targeted therapy in adenoid cystic carcinoma of the head and neck: A review. Head Neck 2010, 33, 905–911. [Google Scholar] [CrossRef]
- Humtsoe, J.O.; Kim, H.-S.; Leonard, B.; Ling, S.; Keam, B.; Marchionni, L.; Afsari, B.; Considine, M.; Favorov, A.V.; Fertig, E.J.; et al. Newly Identified Members of FGFR1 Splice Variants Engage in Cross-talk with AXL/AKT Axis in Salivary Adenoid Cystic Carcinoma. Cancer Res. 2021, 81, 1001–1013. [Google Scholar] [CrossRef]
- Das, S.; Marsden, P.A. Angiogenesis in Glioblastoma. N. Engl. J. Med. 2013, 369, 1561–1563. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Huveldt, D.; Kreinest, P.; Lohse, C.M.; Cheville, J.C.; Parker, A.S.; Copland, J.A.; Anastasiadis, P.Z. A p120 Catenin Isoform Switch Affects Rho Activity, Induces Tumor Cell Invasion, and Predicts Metastatic Disease. J. Biol. Chem. 2008, 283, 18344–18354. [Google Scholar] [CrossRef] [PubMed]
- Marzese, D.M.; Manughian-Peter, A.O.; Orozco, J.I.J.; Hoon, D.S.B. Alternative splicing and cancer metastasis: Prognostic and therapeutic applications. Clin. Exp. Metastasis 2018, 35, 393–402. [Google Scholar] [CrossRef]
- Yuan, F.; Ming, H.; Wang, Y.; Yang, Y.; Yi, L.; Li, T.; Ma, H.; Tong, L.; Zhang, L.; Liu, P.; et al. Molecular and clinical characterization of Galectin-9 in glioma through 1,027 samples. J. Cell. Physiol. 2020, 235, 4326–4334. [Google Scholar] [CrossRef]
- Ferrarese, R.; Harsh, G.R.; Yadav, A.; Bug, E.; Maticzka, D.; Reichardt, W.; Dombrowski, S.M.; Miller, T.E.; Masilamani, A.P.; Dai, F.; et al. Lineage-specific splicing of a brain-enriched alternative exon promotes glioblastoma progression. J. Clin. Investig. 2014, 124, 2861–2876. [Google Scholar] [CrossRef]
- Vashishtha, V.; Jinghan, N.; Yadav, A.K. Antagonistic role of GSK3 isoforms in glioma survival. J. Cancer 2018, 9, 1846–1855. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Fu, W.-M. Identification of differential splicing genes in gliomas using exon expression profiling. Mol. Med. Rep. 2015, 11, 843–850. [Google Scholar] [CrossRef]
- Kuestner, E.R.; Elrod, R.D.; Grant, F.J.; Hagen, F.S.; Kuijper, J.L.; Matthewes, S.L.; O’Hara, P.J.; Sheppard, P.O.; Stroop, S.D.; Thompson, D.L.; et al. Cloning and characterization of an abundant subtype of the human calcitonin receptor. Mol. Pharmacol. 1994, 46, 246–255. [Google Scholar] [PubMed]
- Gilabert-Oriol, R.; Furness, S.G.B.; Stringer, B.W.; Weng, A.; Fuchs, H.; Day, B.W.; Kourakis, A.; Boyd, A.W.; Hare, D.L.; Thakur, M.; et al. Dianthin-30 or gelonin versus monomethyl auristatin E, each configured with an anti-calcitonin receptor antibody, are differentially potent in vitro in high-grade glioma cell lines derived from glioblastoma. Cancer Immunol. Immunother. 2017, 114, 97–1228. [Google Scholar] [CrossRef]
- Ostrovskaya, A.; Hick, C.; Hutchinson, D.S.; Stringer, B.W.; Wookey, P.J.; Wootten, D.; Sexton, P.M.; Furness, S.G.B. Expression and activity of the calcitonin receptor family in a sample of primary human high-grade gliomas. BMC Cancer 2019, 19, 157. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Furness, S.G.B.; Bittencourt, L.; Hare, D.L.; Wookey, P.J. Building the case for the calcitonin receptor as a viable target for the treatment of glioblastoma. Ther. Adv. Med. Oncol. 2020, 12. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.; Threadgill, D.; Eley, G.D.; Strunk, K.E.; Danielsen, A.J.; Sinclair, C.S.; Pearsallb, R.S.; Green, P.J.; Yeec, D.; Lampland, A.L.; et al. Comparative Genomic Sequence Analysis and Isolation of Human and Mouse Alternative EGFR Transcripts Encoding Truncated Receptor Isoforms. Genomics 2001, 71, 1–20. [Google Scholar] [CrossRef]
- Voelzke, W.R.; Petty, W.J.; Lesser, G.J. Targeting the Epidermal Growth Factor Receptor in High-Grade Astrocytomas. Curr. Treat. Options Oncol. 2008, 9, 23–31. [Google Scholar] [CrossRef]
- Albitar, L.; Pickett, G.; Morgan, M.; Wilken, J.A.; Maihle, N.J.; Leslie, K.K. EGFR isoforms and gene regulation in human endometrial cancer cells. Mol. Cancer 2010, 9, 166. [Google Scholar] [CrossRef] [PubMed]
- Guillaudeau, A.; Durand, K.; Rabinovitch-Chable, H.; Pommepuy, I.; Mesturoux, L.; Robert, S.; Chaunavel, A.; Moreau, J.-J.; Labrousse, F. Adult diffuse gliomas produce mRNA transcripts encoding EGFR isoforms lacking a tyrosine kinase domain. Int. J. Oncol. 2011, 40, 1142–1152. [Google Scholar] [CrossRef]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef]
- Sami, A.; Karsy, M. Targeting the PI3K/AKT/mTOR signaling pathway in glioblastoma: Novel therapeutic agents and advances in understanding. Tumor Biol. 2013, 34, 1991–2002. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Zhao, H.F.; Wang, J.; Shao, W.; Wu, C.P.; Chen, Z.P.; To, S.T.; Li, W.P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef] [PubMed]
- Mogilevsky, M.; Shimshon, O.; Kumar, S.; Mogilevsky, A.; Keshet, E.; Yavin, E.; Heyd, F.; Karni, R. Modulation of MKNK2 alternative splicing by splice-switching oligonucleotides as a novel approach for glioblastoma treatment. Nucleic Acids Res. 2018, 46, 11396–11404. [Google Scholar] [CrossRef]
- Maimon, A.; Mogilevsky, M.; Shilo, A.; Golan-Gerstl, R.; Obiedat, A.; Ben-Hur, V.; Lebenthal-Loinger, I.; Stein, I.; Reich, R.; Beenstock, J.; et al. Mnk2 Alternative Splicing Modulates the p38-MAPK Pathway and Impacts Ras-Induced Transformation. Cell Rep. 2014, 7, 501–513. [Google Scholar] [CrossRef]
- Sachamitr, P.; Ho, J.C.; Ciamponi, F.E.; Ba-Alawi, W.; Coutinho, F.J.; Guilhamon, P.; Kushida, M.M.; Cavalli, F.M.G.; Lee, L.; Rastegar, N.; et al. PRMT5 inhibition disrupts splicing and stemness in glioblastoma. Nat. Commun. 2021, 12, 979. [Google Scholar] [CrossRef]
- Li, Y.; Ren, Z.; Peng, Y.; Li, K.; Wang, X.; Huang, G.; Qi, S.; Liu, Y. Classification of glioma based on prognostic alternative splicing. BMC Med. Genom. 2019, 12, 165. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, Y.; Waku, T.; Iwasaki, N.; Ono, W.; Yamaguchi, C.; Yanagisawa, J. Global analysis of DNA methylation in early-stage liver fibrosis. BMC Med. Genom. 2012, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Z.; Zhao, B.; Chen, W.; Wang, Y.; Ma, W. Development of a nomogram for prognostic prediction of lower-grade glioma based on alternative splicing signatures. Cancer Med. 2020, 9, 9266–9281. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Zhang, P.; Wang, X.; Wang, K.; Zhou, M.; Long, H.; Lin, J.; Wu, Z.; Gao, L.; Song, Y. Identification of Prognostic Signatures of Alternative Splicing in Glioma. J. Mol. Neurosci. 2020, 70, 1484–1492. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef]
- van Der Stok, E.P.; Spaander, M.C.W.; Grünhagen, D.J.; Verhoef, C.; Kuipers, E.J. Surveillance after curative treatment for colorectal cancer. Nat. Rev. Clin. Oncol. 2017, 14, 297–315. [Google Scholar] [CrossRef]
- Van Der Jeught, K.; Xu, H.-C.; Li, Y.-J.; Lu, X.-B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef]
- Tanaka, T.; Tanaka, M.; Tanaka, T.; Ishigamori, R. Biomarkers for Colorectal Cancer. Int. J. Mol. Sci. 2010, 11, 3209–3225. [Google Scholar] [CrossRef] [PubMed]
- Litvak, A.; Cercek, A.; Segal, N.; Reidy-Lagunes, D.; Stadler, Z.K.; Yaeger, R.D.; Kemeny, N.E.; Weiser, M.R.; Pessin, M.; Saltz, L. False-Positive Elevations of Carcinoembryonic Antigen in Patients With a History of Resected Colorectal Cancer. J. Natl. Compr. Cancer Netw. 2014, 12, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Pentheroudakis, G.; Mavroeidis, L.; Papadopoulou, K.; Koliou, G.-A.; Bamia, C.; Chatzopoulos, K.; Samantas, E.; Mauri, D.; Efstratiou, I.; Pectasides, D.; et al. Angiogenic and Antiangiogenic VEGFA Splice Variants in Colorectal Cancer: Prospective Retrospective Cohort Study in Patients Treated with Irinotecan-Based Chemotherapy and Bevacizumab. Clin. Color. Cancer 2019, 18, e370–e384. [Google Scholar] [CrossRef]
- Zadeh, M.A.H.; Amin, E.M.; Hoareau-Aveilla, C.; Domingo, E.; Symonds, K.E.; Ye, X.; Heesom, K.J.; Salmon, A.; D’Silva, O.; Betteridge, K.B.; et al. Alternative splicing of TIA-1 in human colon cancer regulates VEGF isoform expression, angiogenesis, tumour growth and bevacizumab resistance. Mol. Oncol. 2014, 9, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, J.M.; Valcarcel, J. Two Isoforms of the T-cell Intracellular Antigen 1 (TIA-1) Splicing Factor Display Distinct Splicing Regulation Activities. J. Biol. Chem. 2007, 282, 19410–19417. [Google Scholar] [CrossRef]
- Ni, B.; Hu, J.; Chen, D.; Li, L.; Chen, D.; Wang, J.; Wang, L. Alternative splicing of spleen tyrosine kinase differentially regulates colorectal cancer progression. Oncol. Lett. 2016, 12, 1737–1744. [Google Scholar] [CrossRef]
- Howlader, N.N.A.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; Chen, H.S.; et al. Cronin KA SEER Cancer Statistics Review, 1975-2018. National Cancer Institute. Bethesda. 2021. Available online: https://seer.cancer.gov/csr/1975_2018/ (accessed on 30 June 2021).
- Dhakal, P.; Lyden, E.; Rajasurya, V.; Zeidan, A.M.; Chaulagain, C.; Gundabolu, K.; Bhatt, V.R. Early mortality and overall survival in acute promyelocytic leukemia: Do real-world data match results of the clinical trials? Leuk. Lymphoma 2021, 1–9. [Google Scholar] [CrossRef]
- Ganzel, C.; Wang, X.V.; Rowe, J.M.; Richards, S.M.; Buck, G.; Marks, D.I.; Litzow, M.R.; Paietta, E.M.; Foroni, L.; Luger, S.M.; et al. At three years, patients with acute lymphoblastic leukaemia are still at risk for relapse. Results of the international MRC UKALLXII/ECOG E2993 trial. Br. J. Haematol. 2020, 191, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef]
- Rivera, O.D.; Mallory, M.J.; Quesnel-Vallières, M.; Chatrikhi, R.; Schultz, D.C.; Carroll, M.; Barash, Y.; Cherry, S.; Lynch, K.W. Alternative splicing redefines landscape of commonly mutated genes in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.N.; Venugopal, P.; Scott, H.; Hiwase, D.K. Splice factor mutations and alternative splicing as drivers of hematopoietic malignancy. Immunol. Rev. 2014, 263, 257–278. [Google Scholar] [CrossRef]
- Sciarrillo, R.; Wojtuszkiewicz, A.; Assaraf, Y.G.; Jansen, G.; Kaspers, G.J.; Giovannetti, E.; Cloos, J. The role of alternative splicing in cancer: From oncogenesis to drug resistance. Drug Resist. Updat. 2020, 53, 100728. [Google Scholar] [CrossRef]
- Rossi, D.; Bruscaggin, A.; Spina, V.; Rasi, S.; Khiabanian, H.; Messina, M.; Fangazio, M.; Vaisitti, T.; Monti, S.; Chiaretti, S.; et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: Association with progression and fludarabine-refractoriness. Blood 2011, 118, 6904–6908. [Google Scholar] [CrossRef]
- Meggendorfer, M.; Roller, A.; Haferlach, T.; Eder, C.; Dicker, F.; Grossmann, V.; Kohlmann, A.; Alpermann, T.; Yoshida, K.; Ogawa, S.; et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood 2012, 120, 3080–3088. [Google Scholar] [CrossRef]
- De Necochea-Campion, R.; Shouse, G.; Zhou, Q.; Mirshahidi, S.; Chen, C.-S. Aberrant splicing and drug resistance in AML. J. Hematol. Oncol. 2016, 9, 85. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Raz, S.; Stark, M.; Assaraf, Y.G. Folylpoly-γ-glutamate synthetase: A key determinant of folate homeostasis and antifolate resistance in cancer. Drug Resist. Updat. 2016, 28, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.; Wichman, C.; Avivi, I.; Assaraf, Y.G. Aberrant splicing of folylpolyglutamate synthetase as a novel mechanism of antifolate resistance in leukemia. Blood 2009, 113, 4362–4369. [Google Scholar] [CrossRef] [PubMed]
- Wojtuszkiewicz, A.; Assaraf, Y.G.; Hoekstra, M.; Sciarrillo, R.; Jansen, G.; Peters, G.J.; Pieters, R.; Sonneveld, E.; Escherich, G.; Kaspers, G.J.; et al. The association of aberrant folylpolyglutamate synthetase splicing with ex vivo methotrexate resistance and clinical outcome in childhood acute lymphoblastic leukemia. Haematologica 2016, 101, e291–e294. [Google Scholar] [CrossRef]
- Wojtuszkiewicz, A.; Raz, S.; Stark, M.; Assaraf, Y.G.; Jansen, G.; Peters, G.J.; Sonneveld, E.; Kaspers, G.J.L.; Cloos, J. Folylpolyglutamate synthetase splicing alterations in acute lymphoblastic leukemia are provoked by methotrexate and other chemotherapeutics and mediate chemoresistance. Int. J. Cancer 2016, 138, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Shahidi, H.; Vottero, A.; Stratakis, C.A.; Taymans, S.E.; Karl, M.; Longui, C.A.; Chrousos, G.P.; Daughaday, W.H.; Gregory, S.A.; Plate, J.M. Imbalanced Expression of the Glucocorticoid Receptor Isoforms in Cultured Lymphocytes from a Patient with Systemic Glucocorticoid Resistance and Chronic Lymphocytic Leukemia. Biochem. Biophys. Res. Commun. 1999, 254, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Longui, C.; Vottero, A.; Adamson, P.; Cole, D.; Kino, T.; Monte, O.; Chrousos, G. Low Glucocorticoid Receptor α/β Ratio in T-cell Lymphoblastic Leukemia. Horm. Metab. Res. 2000, 32, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Matsuzaki, A.; Suminoe, A.; Hattori, H.; Kanemitsu, S.; Hara, T. Differential mRNA expression of glucocorticoid receptor α and β is associated with glucocorticoid sensitivity of acute lymphoblastic leukemia in children. Pediatr. Blood Cancer 2005, 45, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Tuffin, L.J. The Physiology of Human Glucocorticoid Receptor β (hGRbeta) and Glucocorticoid Resistance. Ann. N. Y. Acad. Sci. 2006, 1069, 1–9. [Google Scholar] [CrossRef]
- de Lange, P.; Segeren, C.M.; Koper, J.W.; Wiemer, E.; Sonneveld, P.; Brinkmann, A.O.; White, A.; Brogan, I.J.; De Jong, F.H.; Lamberts, S.W. Expression in hematological malignancies of a glucocorticoid receptor splice variant that augments glucocorticoid receptor-mediated effects in transfected cells. Cancer Res. 2001, 61, 3937–3941. [Google Scholar] [PubMed]
- Moalli, A.P.; Pillay, S.; Krett, N.L.; Rosen, S.T. Alternatively spliced glucocorticoid receptor messenger RNAs in glucocorticoid-resistant human multiple myeloma cells. Cancer Res. 1993, 53, 3877–3879. [Google Scholar]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Raa, G.D.T.; Derks, I.A.M.; Navrkalova, V.; Skowroñska, A.; Moerland, P.D.; Van Laar, J.; Oldreive, C.; Monsuur, H.; Trbusek, M.; Malcikova, J.; et al. The impact of SF3B1 mutations in CLL on the DNA-damage response. Leukemia 2014, 29, 1133–1142. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.A.; Wu, G.; Sussman, R.T.; LaNauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef]
- Chen, S.-L.; Dai, Y.-J.; Hu, F.; Wang, Y.; Li, H.; Liang, Y. Effects of Alternative Splicing Events on Acute Myeloid Leukemia. DNA Cell Biol. 2020, 39, 2040–2051. [Google Scholar] [CrossRef]
- Jin, P.; Tan, Y.; Zhang, W.; Li, J.; Wang, K. Prognostic alternative mRNA splicing signatures and associated splicing factors in acute myeloid leukemia. Neoplasia 2020, 22, 447–457. [Google Scholar] [CrossRef]
- Hershberger, C.E.; Moyer, D.C.; Adema, V.; Kerr, C.M.; Walter, W.; Hutter, S.; Meggendorfer, M.; Baer, C.; Kern, W.; Nadarajah, N.; et al. Complex landscape of alternative splicing in myeloid neoplasms. Leukemia 2021, 35, 1108–1120. [Google Scholar] [CrossRef]
- Mohamed, A.M.; Balsat, M.; Thenoz, M.; Koering, C.; Payen-Gay, L.; Cheok, M.; Mortada, H.; Auboeuf, D.; Pinatel, C.; El-Hamri, M.; et al. Oncogene- and drug resistance-associated alternative exon usage in acute myeloid leukemia (AML). Oncotarget 2015, 7, 2889–2909. [Google Scholar] [CrossRef]
- Veuger, M.J.T.; Heemskerk, M.H.; Honders, M.W.; Willemze, R.; Barge, R.M.Y. Functional role of alternatively spliced deoxycytidine kinase in sensitivity to cytarabine of acute myeloid leukemic cells. Blood 2002, 99, 1373–1380. [Google Scholar] [CrossRef]
- Veuger, M.J.; Honders, M.W.; Landegent, J.E.; Willemze, R.; Barge, R.M. High incidence of alternatively spliced forms of deoxycytidine kinase in patients with resistant acute myeloid leukemia. Blood 2000, 96, 1517–1524. [Google Scholar] [CrossRef]
- Cai, J.; Damaraju, V.L.; Groulx, N.; Mowles, D.; Peng, Y.; Robins, M.J.; Cass, C.E.; Gros, P. Two Distinct Molecular Mechanisms Underlying Cytarabine Resistance in Human Leukemic Cells. Cancer Res. 2008, 68, 2349–2357. [Google Scholar] [CrossRef]
- Stark, M.; Bram, E.; Akerman, M.; Mandel-Gutfreund, Y.; Assaraf, Y.G. Heterogeneous Nuclear Ribonucleoprotein H1/H2-dependent Unsplicing of Thymidine Phosphorylase Results in Anticancer Drug Resistance. J. Biol. Chem. 2011, 286, 3741–3754. [Google Scholar] [CrossRef]
- Goff, D.J.; Recart, A.C.; Sadarangani, A.; Chun, H.-J.; Barrett, C.L.; Krajewska, M.; Leu, H.; Low-Marchelli, J.; Ma, W.; Shih, A.Y.; et al. A Pan-BCL2 Inhibitor Renders Bone-Marrow-Resident Human Leukemia Stem Cells Sensitive to Tyrosine Kinase Inhibition. Cell Stem Cell 2013, 12, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Alonzo, T.A.; Gerbing, R.B.; Lange, B.J.; Heerema, N.A.; Franklin, J.; Raimondi, S.C.; Hirsch, B.A.; Gamis, A.S.; Meshinchi, S. BIRC5 (survivin) splice variant expression correlates with refractory disease and poor outcome in pediatric acute myeloid leukemia: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2013, 61, 647–652. [Google Scholar] [CrossRef]
- Notarbartolo, M.; Cervello, M.; Dusonchet, L.; D’Alessandro, N. NAIP-ΔEx10-11: A novel splice variant of the apoptosis inhibitor NAIP differently expressed in drug-sensitive and multidrug-resistant HL60 leukemia cells. Leuk. Res. 2002, 26, 857–862. [Google Scholar] [CrossRef]
- Ånensen, N.; Hjelle, S.M.; Van Belle, W.; Haaland, I.; Silden, E.; Bourdon, J.-C.; Hovland, R.; Taskén, K.; Knappskog, S.; Lonning, P.E.; et al. Correlation analysis of p53 protein isoforms with NPM1/FLT3 mutations and therapy response in acute myeloid leukemia. Oncogene 2011, 31, 1533–1545. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.-C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Inokuchi, K.; Dan, K. The study for loss of bcl-xs expression as a prognostic factor in acute myeloid leukemia. Leuk. Res. 2002, 26, 1119–1123. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, X.; Lin, C.; Jia, X.; Zhu, H.; Song, J.; Zhang, Y. Noncoding RNAs regulate alternative splicing in Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Xiao, Y.; Ma, J.; Tang, Y.; Tian, B.; Zhang, Y.; Li, X.; Wu, Z.; Yang, D.; Zhou, Y.; et al. Circular RNAs in Cancer: Emerging functions in hallmarks, stemness, resistance and roles as potential biomarkers. Mol. Cancer 2019, 18, 90. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Yang, J.; Li, X.; Liu, D.; Fu, L.; Wang, X. Functions and mechanisms of circular RNAs in cancer radiotherapy and chemotherapy resistance. Mol. Cancer 2020, 19, 58. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Malignancy | Gene | Splice Variant | Mutation Type | Drug Resistance | Biological Function | Reference |
|---|---|---|---|---|---|---|
| NSCLC | PTPMT1 | Exon skipping | SRSF1 target | Radioresistance | Promotes phosphorylation of AMPK PTEN-like mitochondrial phosphatase | [47] |
| BIM | Alternative splicing | SRSF1 | Imatinib | [48] | ||
| U2AF1 | Loss of function | Gemcitabine, glucocorticoids | Target of the splicing factor quaking (QKI) | [39] | ||
| SRSF1 metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) | SRSF1 + ex2 (MCL1L) Alternative 5′SS of exon 2 (BCL-XL) | N/A | Gemcitabine | MNK2 splicing; MCL1 member of BCL2 family splicing to MCL1L long anti-apoptotic variant | [41,42] | |
| LUAD | EIF4H | EIF4H + ex5 | Gain of function | Resistance to PI(3)K/AKT/mTOR inhibitors (e.g., AZD8055, BEZ235) | Cancer related genes translation | [49] |
| Malignancy | Gene | Splice Variant | Drug Resistance | Biological Action | Reference |
|---|---|---|---|---|---|
| Breast Cancer | HER2 | P100 of HER2 Δ16-HER2 d16HER2 | Trastuzumab | SRSF3 and hnRNPH1 are associated with splicing regulation of Δ16-HER2. d16HER2 influences tumor initiation and aggressiveness, cancer stem cell properties, epithelial-mesenchymal transition and HER2-positive breast cancer cell susceptibility to trastuzumab | [55,57] |
| ECT2 | ECT2-Ex5+ | Doxorubicin | ZRANB2 and SYF2-mediated AS programs converging on ECT2 act in drug resistance | [60] |
| Malignancy | Gene | Splice Variant | Mutation Type | Drug Resistance | Biological Function | Reference |
|---|---|---|---|---|---|---|
| Prostate Cancer | AR | AR-V7 | Cryptique exon 3 inclusion, exon skip | ADT resistance—Enzalutamide, Abiraterone | Activate target gene expression | [84,85] |
| SRSF3 SF3B2 U2AF2 | Alternative splicing | Poison exon insertion | AR-V7 induction | Myc interaction and NEPC induction | [90,102,104] | |
| EZH2 | EZH2 + ex14 | Exon 14 inclusion (promoted by SF3B3) | AR induction | Enzalutamide | [115,119] |
| Malignancy | Gene | Splice Variant | Mutation Type | Drug Resistance | Biological Function | Reference |
|---|---|---|---|---|---|---|
| Adenoid cystic carcinoma (HNSCC) | FGFR1 | FGFR1v | Premature termination codon at position 147 of intronic segment | Dovitinib | Mediate FGF/FGFR1-independent function through the AXL/AKT signaling axis | [149] |
| OSCC | SRSF3 | SRSF3ex4 | Long isoform with exon 4 encodes a truncated SRSF3 protein | Paclitaxel | Increases the expression of c-Jun, cyclin D1, cyclin D3, CDC25A and E2F1, and accelerates cell growth | [58] |
| Malignancy | Gene | Splice Variant | Mutation Type | Drug Resistance | Biological Function | Reference |
|---|---|---|---|---|---|---|
| mCRC | VEGFA | VEGFA145b | Differential splicing of the 3′ distal site of exon 8 | Bevacizumab | Act as a reservoir of angiogenic growth factors in the tumor stroma | [183] |
| TIA-1 | sTIA-1 flTIA-1 | Exon 5 exclusion leading to truncated protein | Anti-VEGF antibodies | Alters both co-transcriptional and post-transcriptional RNA processing | [184] |
| Malignancy | Gene | Splice Variant | Mutation Type | Drug Resistance | Biological Function | Reference |
|---|---|---|---|---|---|---|
| CLL | SF3B1 | SF3B1-ΔHEAT | Deletions in HEAT domains | Fludarabine | Splicosome factor | [194] |
| T-ALL, cALL (both T- and B-cell) | FPGS | FPGS-ES(12);IR(8) | Exon 12 skipping; intron 8 partial retention | Methotrexate Dexamethasone, Mitoxanthrone, Prednisolone | Intracellular modification of MTX | [198,199,200] |
| ALL | GR | GRβ | Downstream acceptor site in exon 9 | Glucocorticoids | Inactive GC receptor; dominant negative isoform | [38] |
| P53 | P53β | Exon 9β inclusion | Glucocorticoids | Higher expression in resistant cells | [39] | |
| AML | dCT | dCT-ΔEx2–6 | Missing exons 2–6 (deletions) | Cytarabine | Enzyme, which activates Cytarabine | [214,215] |
| TET2 | TET2-ES(2) | Skipping of exon 2 | Cytarabine | [213] | ||
| P53 | P53β/γ | Alternative splicing of exon 9β or 9γ | Doxorubicin | Better prognosis/active tumor suppressor | [221] | |
| Bcl-x | Bcl-xs | Alternative splicing | Multiple drugs | Altered apoptosis; loss of bcl-xs leads to worse RFS and OS | [223] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehterov, N.; Kazakova, M.; Sbirkov, Y.; Vladimirov, B.; Belev, N.; Yaneva, G.; Todorova, K.; Hayrabedyan, S.; Sarafian, V. Alternative RNA Splicing—The Trojan Horse of Cancer Cells in Chemotherapy. Genes 2021, 12, 1085. https://doi.org/10.3390/genes12071085
Mehterov N, Kazakova M, Sbirkov Y, Vladimirov B, Belev N, Yaneva G, Todorova K, Hayrabedyan S, Sarafian V. Alternative RNA Splicing—The Trojan Horse of Cancer Cells in Chemotherapy. Genes. 2021; 12(7):1085. https://doi.org/10.3390/genes12071085
Chicago/Turabian StyleMehterov, Nikolay, Maria Kazakova, Yordan Sbirkov, Boyan Vladimirov, Nikolay Belev, Galina Yaneva, Krassimira Todorova, Soren Hayrabedyan, and Victoria Sarafian. 2021. "Alternative RNA Splicing—The Trojan Horse of Cancer Cells in Chemotherapy" Genes 12, no. 7: 1085. https://doi.org/10.3390/genes12071085
APA StyleMehterov, N., Kazakova, M., Sbirkov, Y., Vladimirov, B., Belev, N., Yaneva, G., Todorova, K., Hayrabedyan, S., & Sarafian, V. (2021). Alternative RNA Splicing—The Trojan Horse of Cancer Cells in Chemotherapy. Genes, 12(7), 1085. https://doi.org/10.3390/genes12071085