
Prenatal Versus Postnatal Diagnosis of Meckel–Gruber and Joubert Syndrome in Patients with TMEM67 Mutations

 , , , and
, , , and
Abstract
:1. Introduction
2. Clinical Reports
2.1. Case #1
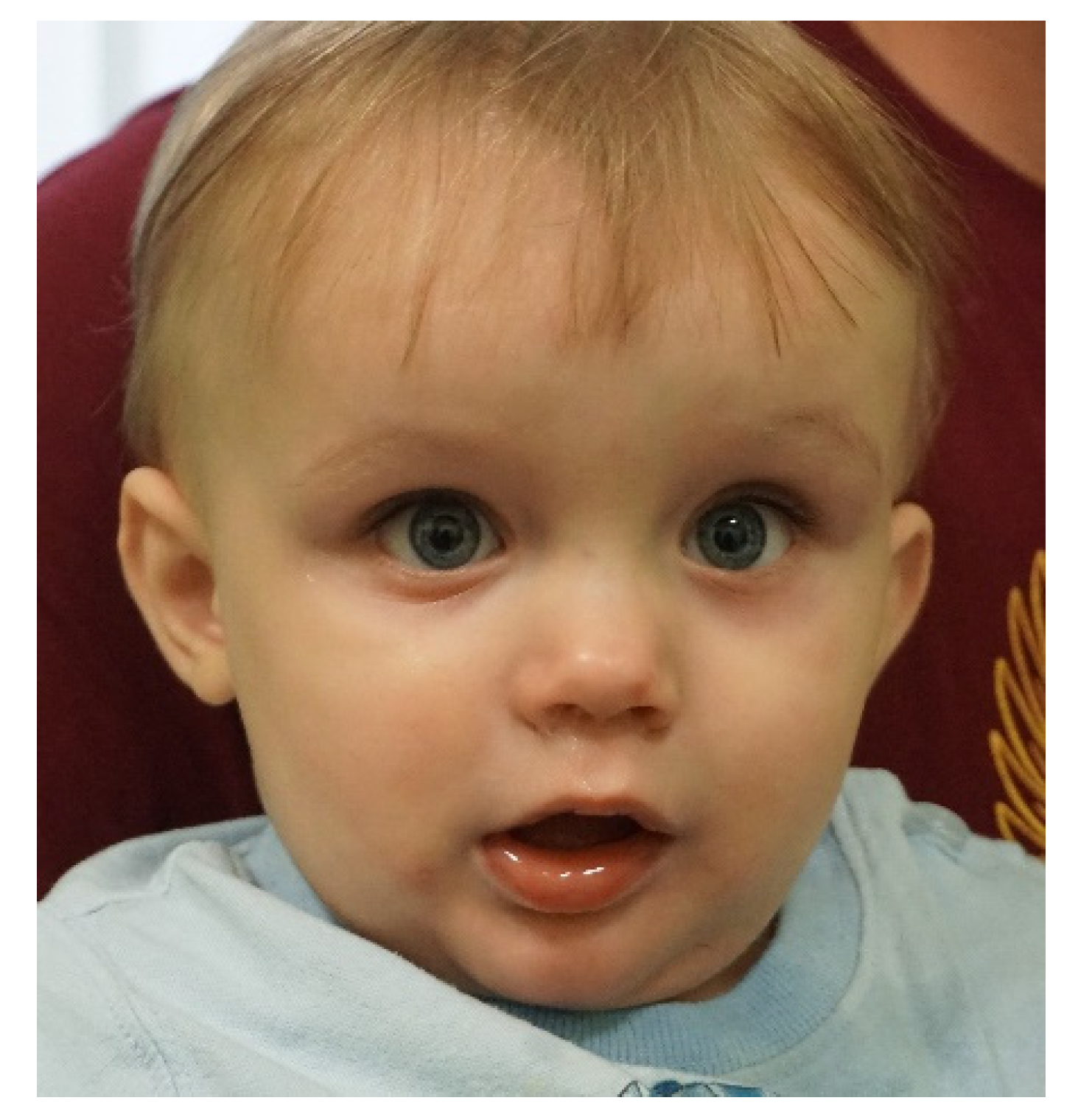
2.2. Case #2
3. Methods of Genetic Analyses
4. Results of Genetic Tests
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferro, F.; Vezzali, N.; Comploj, E.; Pedron, E.; Di Serafino, M.; Esposito, F.; Pelliccia, P.; Rossi, E.; Zeccolini, M.; Gianfranco, V. Pediatric Cystic Diseases of the Kidney. J. Ultrasound 2019, 22, 381–393. [Google Scholar] [CrossRef]
- Gimpel, C.; Avni, F.E.; Bergmann, C.; Cetiner, M.; Habbig, S.; Haffner, D.; König, J.; Konrad, M.; Liebau, M.C.; Pape, L.; et al. Perinatal Diagnosis, Management, and Follow-up of Cystic Renal Diseases. A Clinical Practice Recommendation with Systematic Literature Reviews. JAMA Pediatr. 2018, 172, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Sigmon, D.F.; Shikhman, R.; Nielson, J.I. Renal cyst. StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Khare, A.; Krishnappa, V.; Kumar, D.; Raina, R. Neonatal renal cystic diseases. J. Matern. Fetal. Neonatal. Med. 2018, 31, 2923–2929. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, T.; Makabe, S.; Aoyama, Y.; Kataoka, H.; Nitta, K. New insights into cystic kidney diseases. Contrib. Nephrol. 2018, 195, 31–41. [Google Scholar]
- Veldman, T.; Vignon, C.; Schröck, E.; Rowley, J.D.; Ried, T. Hidden chromosome abnormalities in haematological malignancies detected by multicolour spectral karyotyping. Nat. Genet. 1997, 15, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Smigiel, R.; Biela, M.; Szmyd, K.; Bloch, M.; Szmida, E.; Skiba, P.; Walczak, A.; Gasperowicz, P.; Kosinska, J.; Rydzanicz, M.; et al. Rapid Whole-Exome Sequencing as a Diagnostic Tool in a Neonatal/Pediatric Intensive Care Unit. J. Clin. Med. 2020, 9, 2220. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef]
- Kagan, K.O.; Dufke, A.; Gembruch, U. Renal cystic disease and associated ciliopathies. Curr. Opin. Obstet. Gynecol. 2017, 29, 85–94. [Google Scholar] [CrossRef]
- Bruechle, N.O.; Steuernagel, P.; Zerres, K.; Kurth, I.; Eggermann, T.; Knopp, C. Uniparental disomy as an unexpected cause of Meckel-Gruber syndrome: Report of a case. Pediatr. Nephrol. 2017, 32, 1989–1992. [Google Scholar] [CrossRef]
- Parisi, M.A. The molecular genetics of Joubert syndrome and related ciliopathies: The challenges of genetic and phenotypic heterogeneity. Transl. Sci. Rare Dis. 2019, 4, 25–49. [Google Scholar] [CrossRef]
- Barker, A.R.; Thomas, R.; Dawe, H.R. Meckel-Gruber syndrome and the role of primary cilia in kidney, skeleton, and central nervous system development. Organogenesis 2014, 10, 96–107. [Google Scholar] [CrossRef]
- Hartill, V.; Szymanska, K.; Sharif, S.M.; Wheway, G.; Johnson, C.A. Meckel–Gruber Syndrome: An Update on Diagnosis, Clinical Management, and Research Advances. Front. Pediatr. 2017, 5, 244. [Google Scholar] [CrossRef]
- Barisic, I.; Boban, L.; Loane, M.; Garne, E.; Wellesley, D.; Calzolari, E.; Dolk, H.; Addor, M.C.; Bergman, J.E.; Braz, P.; et al. Meckel-Gruber Syndrome: A Population-Based Study on Prevalence, Prenatal Diagnosis, Clinical Features, and Survival in Europe. Eur. J. Hum. Genet. 2015, 23, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, U.; Malla, T.; Joshi, K.S. Meckel-Gruber syndrome. Kathmandu Univ Med. J. KUMJ 2006, 4, 334–336. [Google Scholar] [PubMed]
- Lannicelli, M.; Brancati, F.; Mougou-Zerelli, S.; Mazzotta, A.; Thomas, S.; Elkhartoufi, N.; Travaglini, L.; Gomes, C.; Ardissino, G.L.; Bertini, E.; et al. Novel TMEM67 mutations and genotype-phenotype correlates in meckelin-related ciliopathies. Hum. Mutat. 2010, 31, E1319–E1331. [Google Scholar]
- Alam, A.; Adhi, M.; Bano, R.; Zubair, A.; Mushtaq, A. Meckel Gruber Syndrome: Second trimester diagnosis of a case in a non-consanguineous marriage. Pak. J. Med. Sci. 2013, 29, 234–236. [Google Scholar]
- Zhu, L.; Xie, L. Prenatal diagnosis of Joubert syndrome. Medicine 2017, 96, e8626. [Google Scholar] [CrossRef]
- Bui, T.P.H.; Nguyen, N.T.; Ngo, V.D.; Nguyen, H.N.; Ly, T.T.H.; Do, H.D.; Huynh, M.T. Novel Compound Heterozygous TMEM67 Variants in a Vietnamese Family With Joubert Syndrome: A Case Report. BMC Med. Genet. 2020, 21, 18. [Google Scholar] [CrossRef]
- Brancati, F.; Dallapiccola, B.; Valente, E.M. Joubert Syndrome and Related Disorders. Orphanet. J. Rare Dis. 2010, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Boycott, K.M.; Parboosingh, J.S.; Scott, J.N.; McLeod, D.R.; Greenberg, C.R.; Fujiwara, T.M.; Mah, J.K.; Midgley, J.; Wade, A.; Bernier, F.P.; et al. Meckel syndrome in the Hutterite population is actually a Joubert-related cerebello-oculo-renal syndrome. Am. J. Med. Genet. A 2007, 143A, 1715–1725. [Google Scholar] [CrossRef]
- Doherty, D. Joubert Syndrome: Insights into Brain Development, Cilium Biology, and Complex Disease. Semin Pediatr. Neurol. 2009, 16, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Doherty DParisi, M.A.; Finn, L.S.; Gunay-Aygun, M.; Al-Mateen, M.; Bates, D.; Clericuzio, C.; Demir, H.; Dorschner, M.; van Essen, A.J.; Gahl, W.A.; et al. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J. Med. Genet. 2010, 47, 8–21. [Google Scholar] [CrossRef]
- Otto, E.A.; Tory, K.; Attanasio, M.; Zhou, W.; Chaki, M.; Paruchuri, Y.; Wise, E.L.; Wolf, M.T.; Utsch, B.; Becker, C.; et al. Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11). J. Med. Genet. 2009, 46, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Szymanska, K.; Jensen, V.L.; Janecke, A.R.; Innes, A.M.; Davis, E.E.; Frosk, P.; Li, C.; Willer, J.R.; Chodirker, B.N.; et al. TMEM237 Is Mutated in Individuals With a Joubert Syndrome Related Disorder and Expands the Role of the TMEM Family at the Ciliary Transition Zone. Am. J. Hum. Genet. 2011, 89, 713–730. [Google Scholar] [CrossRef]
- Huynh, J.M.; Galindo, M.; Laukaitis, C.M. Missense variants in TMEM67 in a patients with Joubert syndrome. Clin. Case Rep. 2018, 6, 2189–2192. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Nothwang, H.G.; Vossmerbäumer, U.; Springer, C.; Strahm, B.; Hoppe, B.; Keuth, B.; Fuchshuber, A.; Querfeld, U.; Neuhaus, T.J.; et al. Lack of Large, Homozygous Deletions of the Nephronophthisis 1 Region in Joubert Syndrome Type B. APN Study Group. Arbeitsgemeinschaft für Pädiatrische Nephrologie. Pediatr. Nephrol. 1998, 12, 16–19. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Patient. | #1 | #2. | ||
|---|---|---|---|---|
| Gene | TMEM67 (NM_153704.5) | |||
| Variant (GRCh38/hg38) | chr8:093795970-T > C c.1843T > C; p. (Cys615Arg) | chr8:093797200-C > T c.1927C > T; p. (Arg643Ter) | chr8:093780615-G > A c.737G > A; p. (Cys246Tyr) | chr8:093797345-C > T c.1975C > T; p. (Arg659Ter) |
| Parental origin | paternal | maternal | maternal | paternal |
| SNP association number (Rsid) | rs201893408 | rs115195998 | no applicable | rs150332116 |
| Frequency a | 0.00009194 | 0.00001315 | 0.0 | 0.00003945 |
| ACMG pathogenicity prediction b | pathogenic | pathogenic | likely pathogenic | pathogenic |
| ClinVar c | Pathogenic/likely pathogenic | no data | no data | no data |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stembalska, A.; Rydzanicz, M.; Pollak, A.; Kostrzewa, G.; Stawinski, P.; Biela, M.; Ploski, R.; Smigiel, R. Prenatal Versus Postnatal Diagnosis of Meckel–Gruber and Joubert Syndrome in Patients with TMEM67 Mutations. Genes 2021, 12, 1078. https://doi.org/10.3390/genes12071078
Stembalska A, Rydzanicz M, Pollak A, Kostrzewa G, Stawinski P, Biela M, Ploski R, Smigiel R. Prenatal Versus Postnatal Diagnosis of Meckel–Gruber and Joubert Syndrome in Patients with TMEM67 Mutations. Genes. 2021; 12(7):1078. https://doi.org/10.3390/genes12071078
Chicago/Turabian StyleStembalska, Agnieszka, Małgorzata Rydzanicz, Agnieszka Pollak, Grazyna Kostrzewa, Piotr Stawinski, Mateusz Biela, Rafal Ploski, and Robert Smigiel. 2021. "Prenatal Versus Postnatal Diagnosis of Meckel–Gruber and Joubert Syndrome in Patients with TMEM67 Mutations" Genes 12, no. 7: 1078. https://doi.org/10.3390/genes12071078
APA StyleStembalska, A., Rydzanicz, M., Pollak, A., Kostrzewa, G., Stawinski, P., Biela, M., Ploski, R., & Smigiel, R. (2021). Prenatal Versus Postnatal Diagnosis of Meckel–Gruber and Joubert Syndrome in Patients with TMEM67 Mutations. Genes, 12(7), 1078. https://doi.org/10.3390/genes12071078